

Structural basis for SHOC2 modulation of RAS signalling
- PMID: 35768504
- PMCID: PMC9452301
- DOI: 10.1038/s41586-022-04838-3
Structural basis for SHOC2 modulation of RAS signalling
Abstract
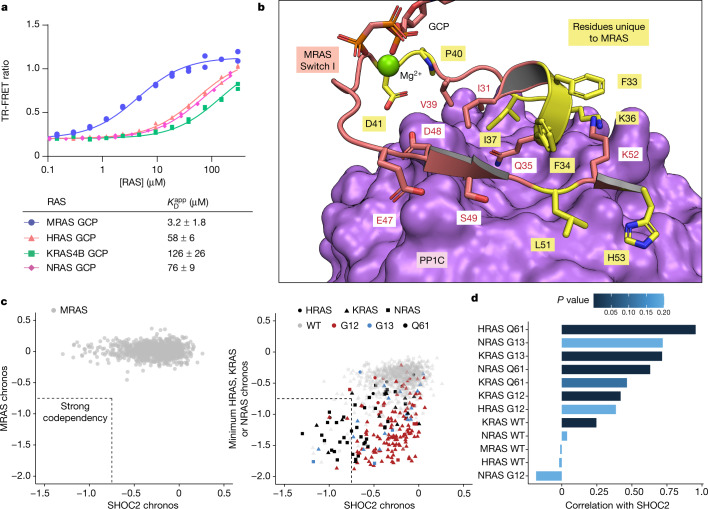
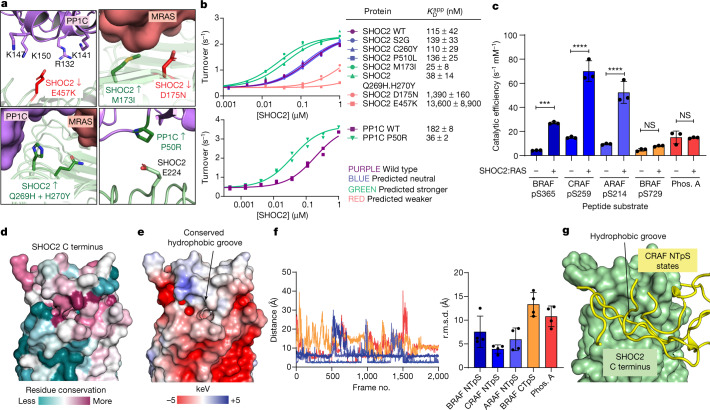
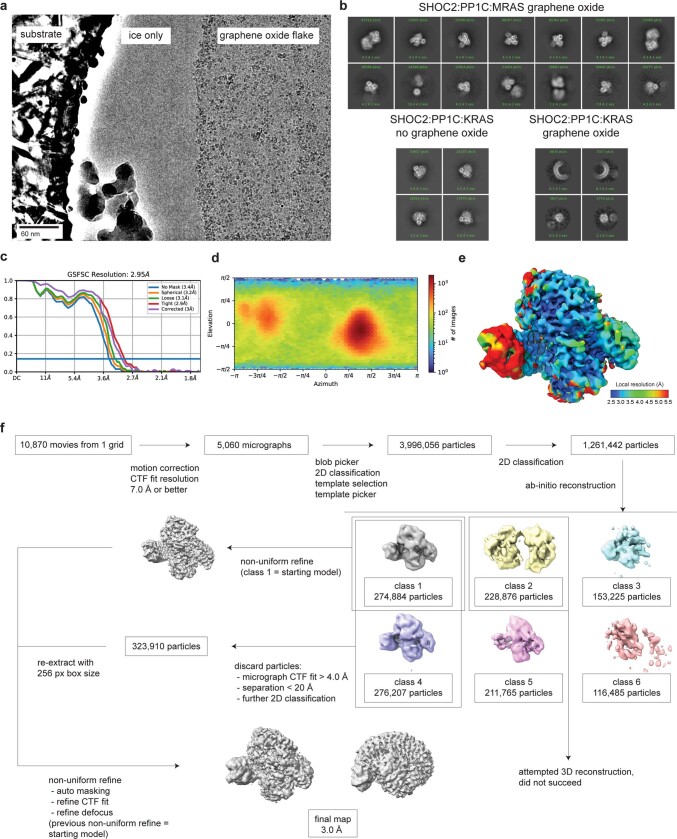
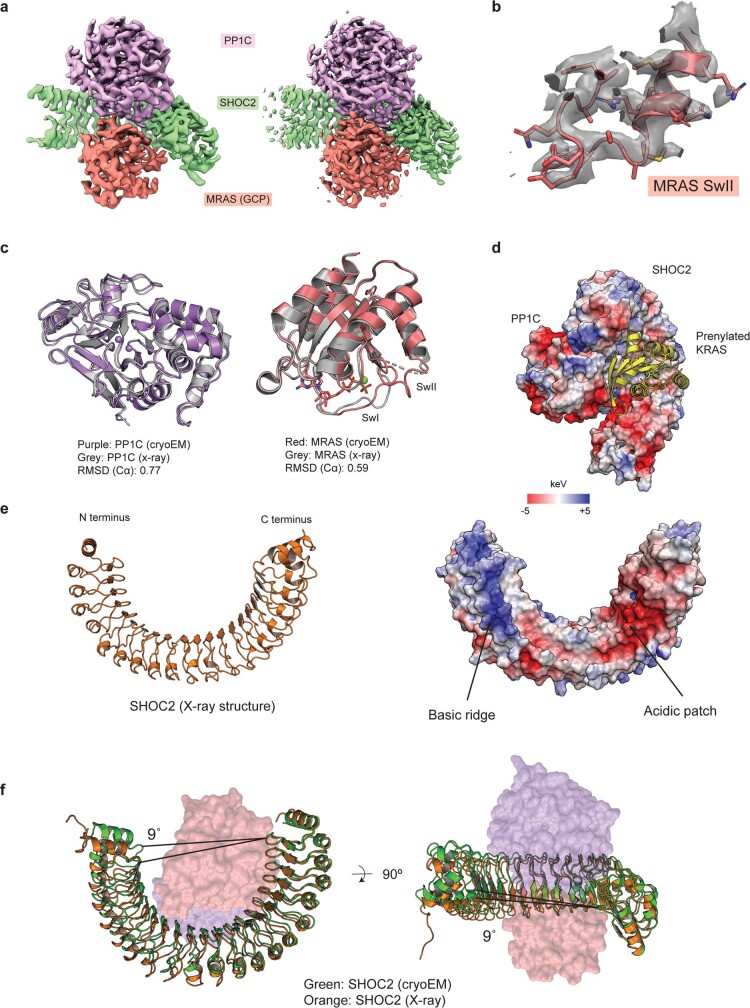
The RAS-RAF pathway is one of the most commonly dysregulated in human cancers1-3. Despite decades of study, understanding of the molecular mechanisms underlying dimerization and activation4 of the kinase RAF remains limited. Recent structures of inactive RAF monomer5 and active RAF dimer5-8 bound to 14-3-39,10 have revealed the mechanisms by which 14-3-3 stabilizes both RAF conformations via specific phosphoserine residues. Prior to RAF dimerization, the protein phosphatase 1 catalytic subunit (PP1C) must dephosphorylate the N-terminal phosphoserine (NTpS) of RAF11 to relieve inhibition by 14-3-3, although PP1C in isolation lacks intrinsic substrate selectivity. SHOC2 is as an essential scaffolding protein that engages both PP1C and RAS to dephosphorylate RAF NTpS11-13, but the structure of SHOC2 and the architecture of the presumptive SHOC2-PP1C-RAS complex remain unknown. Here we present a cryo-electron microscopy structure of the SHOC2-PP1C-MRAS complex to an overall resolution of 3 Å, revealing a tripartite molecular architecture in which a crescent-shaped SHOC2 acts as a cradle and brings together PP1C and MRAS. Our work demonstrates the GTP dependence of multiple RAS isoforms for complex formation, delineates the RAS-isoform preference for complex assembly, and uncovers how the SHOC2 scaffold and RAS collectively drive specificity of PP1C for RAF NTpS. Our data indicate that disease-relevant mutations affect complex assembly, reveal the simultaneous requirement of two RAS molecules for RAF activation, and establish rational avenues for discovery of new classes of inhibitors to target this pathway.
© 2022. The Author(s).
Conflict of interest statement
N.P.D.L., M.J., L.G., S.I., J.M.B., T.J.W., W.P. and J.S. are all employees of Genentech.
Figures









Comment in
-
Structural keys unlock RAS-MAPK cellular signalling pathway.Nature. 2022 Sep;609(7926):248-249. doi: 10.1038/d41586-022-02189-7. Nature. 2022. PMID: 35970881 No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
