

Coronaviruses in wild animals sampled in and around Wuhan at the beginning of COVID-19 emergence
- PMID: 35769892
- PMCID: PMC9214087
- DOI: 10.1093/ve/veac046
Coronaviruses in wild animals sampled in and around Wuhan at the beginning of COVID-19 emergence
Abstract
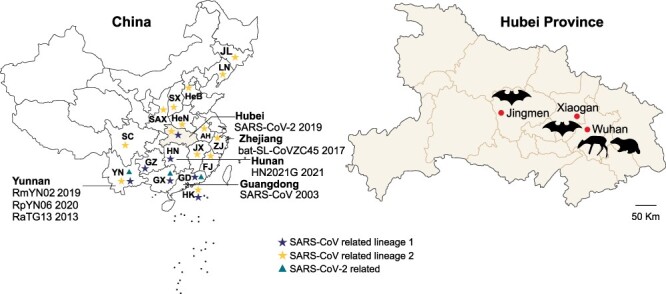
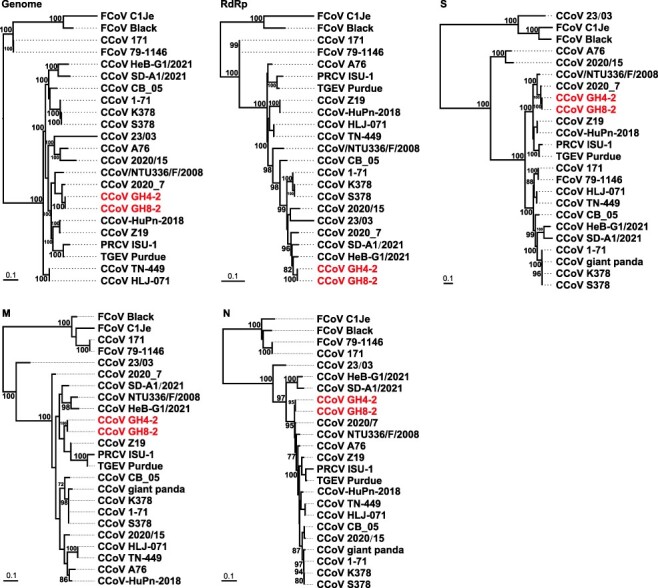
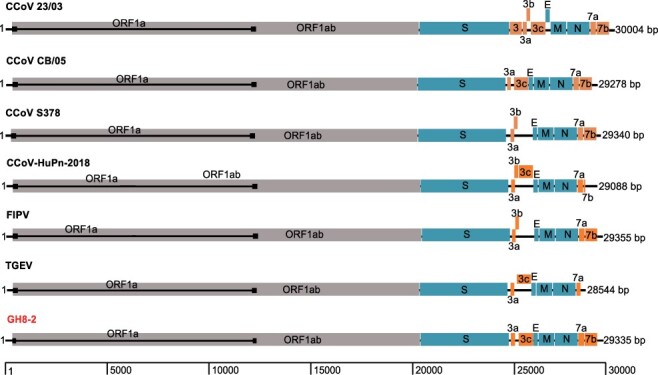
Over the last several decades, no emerging virus has had a profound impact on the world as the SARS-CoV-2 that emerged at the end of 2019 has done. To know where severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) originated from and how it jumped into human population, we immediately started a surveillance investigation in wild mammals in and around Wuhan when we determined the agent. Herein, coronaviruses were screened in the lung, liver, and intestinal tissue samples from fifteen raccoon dogs, seven Siberian weasels, three hog badgers, and three Reeves's muntjacs collected in Wuhan and 334 bats collected around Wuhan. Consequently, eight alphacoronaviruses were identified in raccoon dogs, while nine betacoronaviruses were found in bats. Notably, the newly discovered alphacoronaviruses shared a high whole-genome sequence similarity (97.9 per cent) with the canine coronavirus (CCoV) strain 2020/7 sampled from domestic dog in the UK. Some betacoronaviruses identified here were closely related to previously known bat SARS-CoV-related viruses sampled from Hubei province and its neighbors, while the remaining betacoronaviruses exhibited a close evolutionary relationship with SARS-CoV-related bat viruses in the RdRp gene tree and clustered together with SARS-CoV-2-related bat coronaviruses in the M, N and S gene trees, but with relatively low similarity. Additionally, these newly discovered betacoronaviruses seem unlikely to bind angiotensin-converting enzyme 2 because of the deletions in the two key regions of their receptor-binding motifs. Finally, we did not find SARS-CoV-2 or its progenitor virus in these animal samples. Due to the high circulation of CCoVs in raccoon dogs in Wuhan, more scientific efforts are warranted to better understand their diversity and evolution in China and the possibility of a potential human agent.
Keywords: CCoV; SARS-related coronavirus; Wuhan; bats; raccoon dog.
© The Author(s) 2022. Published by Oxford University Press.
Figures

Similar articles
-
Identification of diverse alphacoronaviruses and genomic characterization of a novel severe acute respiratory syndrome-like coronavirus from bats in China.J Virol. 2014 Jun;88(12):7070-82. doi: 10.1128/JVI.00631-14. Epub 2014 Apr 9. J Virol. 2014. PMID: 24719429 Free PMC article.
-
Extensive diversity of coronaviruses in bats from China.Virology. 2017 Jul;507:1-10. doi: 10.1016/j.virol.2017.03.019. Epub 2017 Apr 3. Virology. 2017. PMID: 28384506 Free PMC article.
-
[Etiology of epidemic outbreaks COVID-19 on Wuhan, Hubei province, Chinese People Republic associated with 2019-nCoV (Nidovirales, Coronaviridae, Coronavirinae, Betacoronavirus, Subgenus Sarbecovirus): lessons of SARS-CoV outbreak.].Vopr Virusol. 2020;65(1):6-15. doi: 10.36233/0507-4088-2020-65-1-6-15. Vopr Virusol. 2020. PMID: 32496715 Review. Russian.
-
[Source of the COVID-19 pandemic: ecology and genetics of coronaviruses (Betacoronavirus: Coronaviridae) SARS-CoV, SARS-CoV-2 (subgenus Sarbecovirus), and MERS-CoV (subgenus Merbecovirus).].Vopr Virusol. 2020;65(2):62-70. doi: 10.36233/0507-4088-2020-65-2-62-70. Vopr Virusol. 2020. PMID: 32515561 Review. Russian.
-
Understanding evolution of SARS-CoV-2: A perspective from analysis of genetic diversity of RdRp gene.J Med Virol. 2020 Oct;92(10):1932-1937. doi: 10.1002/jmv.25909. Epub 2020 Jun 2. J Med Virol. 2020. PMID: 32314811 Free PMC article.
Cited by
-
Genetic tracing of market wildlife and viruses at the epicenter of the COVID-19 pandemic.bioRxiv [Preprint]. 2023 Sep 14:2023.09.13.557637. doi: 10.1101/2023.09.13.557637. bioRxiv. 2023. Update in: Cell. 2024 Sep 19;187(19):5468-5482.e11. doi: 10.1016/j.cell.2024.08.010. PMID: 37745602 Free PMC article. Updated. Preprint.
-
Receptor binding and structural basis of raccoon dog ACE2 binding to SARS-CoV-2 prototype and its variants.PLoS Pathog. 2024 Dec 5;20(12):e1012713. doi: 10.1371/journal.ppat.1012713. eCollection 2024 Dec. PLoS Pathog. 2024. PMID: 39637248 Free PMC article.
-
Emerging Variants of Canine Enteric Coronavirus Associated with Outbreaks of Gastroenteric Disease.Emerg Infect Dis. 2024 Jun;30(6):1240-1244. doi: 10.3201/eid3006.231184. Emerg Infect Dis. 2024. PMID: 38782018 Free PMC article.
-
Lack of detection of SARS-CoV-2 in British wildlife 2020-21 and first description of a stoat (Mustela erminea) Minacovirus.J Gen Virol. 2023 Dec;104(12):001917. doi: 10.1099/jgv.0.001917. J Gen Virol. 2023. PMID: 38059490 Free PMC article.
-
Back to Science in Searching for SARS-CoV-2 Origins.China CDC Wkly. 2023 Apr 7;5(14):315-317. doi: 10.46234/ccdcw2023.055. China CDC Wkly. 2023. PMID: 37193308 Free PMC article. No abstract available.
References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
