

Coordinated Transcriptional and Catabolic Programs Support Iron-Dependent Adaptation to RAS-MAPK Pathway Inhibition in Pancreatic Cancer
- PMID: 35771494
- PMCID: PMC9444964
- DOI: 10.1158/2159-8290.CD-22-0044
Coordinated Transcriptional and Catabolic Programs Support Iron-Dependent Adaptation to RAS-MAPK Pathway Inhibition in Pancreatic Cancer
Abstract
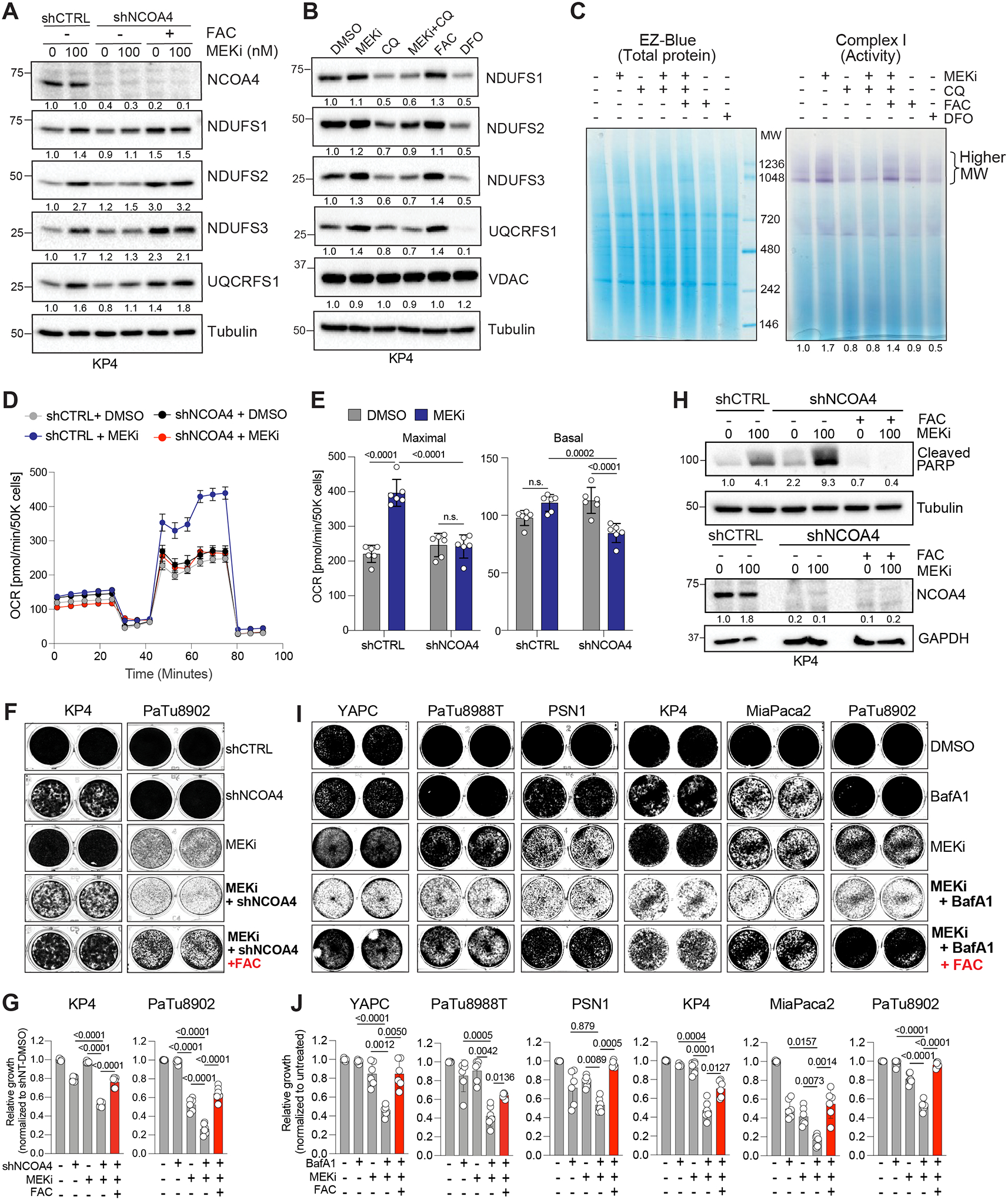
The mechanisms underlying metabolic adaptation of pancreatic ductal adenocarcinoma (PDA) cells to pharmacologic inhibition of RAS-MAPK signaling are largely unknown. Using transcriptome and chromatin immunoprecipitation profiling of PDA cells treated with the MEK inhibitor (MEKi) trametinib, we identify transcriptional antagonism between c-MYC and the master transcription factors for lysosome gene expression, the MiT/TFE proteins. Under baseline conditions, c-MYC and MiT/TFE factors compete for binding to lysosome gene promoters to fine-tune gene expression. Treatment of PDA cells or patient organoids with MEKi leads to c-MYC downregulation and increased MiT/TFE-dependent lysosome biogenesis. Quantitative proteomics of immunopurified lysosomes uncovered reliance on ferritinophagy, the selective degradation of the iron storage complex ferritin, in MEKi-treated cells. Ferritinophagy promotes mitochondrial iron-sulfur cluster protein synthesis and enhanced mitochondrial respiration. Accordingly, suppressing iron utilization sensitizes PDA cells to MEKi, highlighting a critical and targetable reliance on lysosome-dependent iron supply during adaptation to KRAS-MAPK inhibition.
Significance: Reduced c-MYC levels following MAPK pathway suppression facilitate the upregulation of autophagy and lysosome biogenesis. Increased autophagy-lysosome activity is required for increased ferritinophagy-mediated iron supply, which supports mitochondrial respiration under therapy stress. Disruption of ferritinophagy synergizes with KRAS-MAPK inhibition and blocks PDA growth, thus highlighting a key targetable metabolic dependency. See related commentary by Jain and Amaravadi, p. 2023. See related article by Santana-Codina et al., p. 2180. This article is highlighted in the In This Issue feature, p. 2007.
©2022 American Association for Cancer Research.
Figures






Comment in
-
NCOA4-Mediated Ferritinophagy Is a Pancreatic Cancer Dependency via Maintenance of Iron Bioavailability for Iron-Sulfur Cluster Proteins.Cancer Discov. 2022 Sep 2;12(9):2180-2197. doi: 10.1158/2159-8290.CD-22-0043. Cancer Discov. 2022. PMID: 35771492 Free PMC article.
-
Pumping Iron: Ferritinophagy Promotes Survival and Therapy Resistance in Pancreatic Cancer.Cancer Discov. 2022 Sep 2;12(9):2023-2025. doi: 10.1158/2159-8290.CD-22-0734. Cancer Discov. 2022. PMID: 36052501 Free PMC article.
Comment on
-
NCOA4-Mediated Ferritinophagy Is a Pancreatic Cancer Dependency via Maintenance of Iron Bioavailability for Iron-Sulfur Cluster Proteins.Cancer Discov. 2022 Sep 2;12(9):2180-2197. doi: 10.1158/2159-8290.CD-22-0043. Cancer Discov. 2022. PMID: 35771492 Free PMC article.
References
-
- Papke B, Der CJ. Drugging RAS: Know the enemy. Science 2017;355: 1158–63. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
