

Pyruvate and uridine rescue the metabolic profile of OXPHOS dysfunction
- PMID: 35772644
- PMCID: PMC9287363
- DOI: 10.1016/j.molmet.2022.101537
Pyruvate and uridine rescue the metabolic profile of OXPHOS dysfunction
Abstract
Introduction: Primary mitochondrial diseases (PMD) are a large, heterogeneous group of genetic disorders affecting mitochondrial function, mostly by disrupting the oxidative phosphorylation (OXPHOS) system. Understanding the cellular metabolic re-wiring occurring in PMD is crucial for the development of novel diagnostic tools and treatments, as PMD are often complex to diagnose and most of them currently have no effective therapy.
Objectives: To characterize the cellular metabolic consequences of OXPHOS dysfunction and based on the metabolic signature, to design new diagnostic and therapeutic strategies.
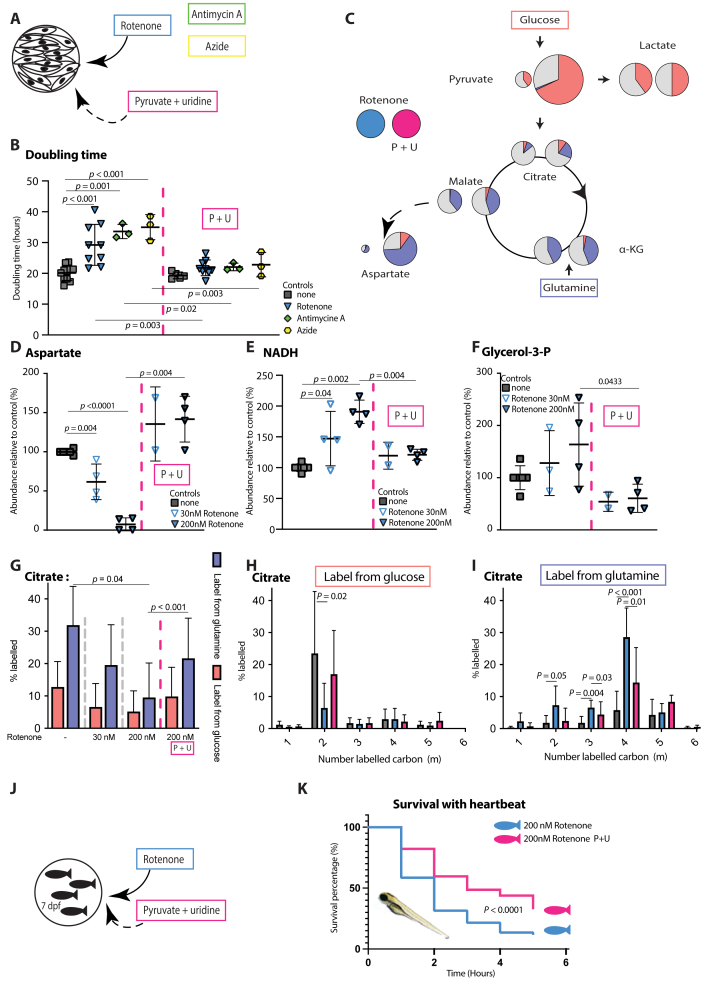
Methods: In vitro assays were performed in skin-derived fibroblasts obtained from patients with diverse PMD and validated in pharmacological models of OXPHOS dysfunction. Proliferation was assessed using the Incucyte technology. Steady-state glucose and glutamine tracing studies were performed with LC-MS quantification of cellular metabolites. The therapeutic potential of nutritional supplements was evaluated by assessing their effect on proliferation and on the metabolomics profile. Successful therapies were then tested in a in vivo lethal rotenone model in zebrafish.
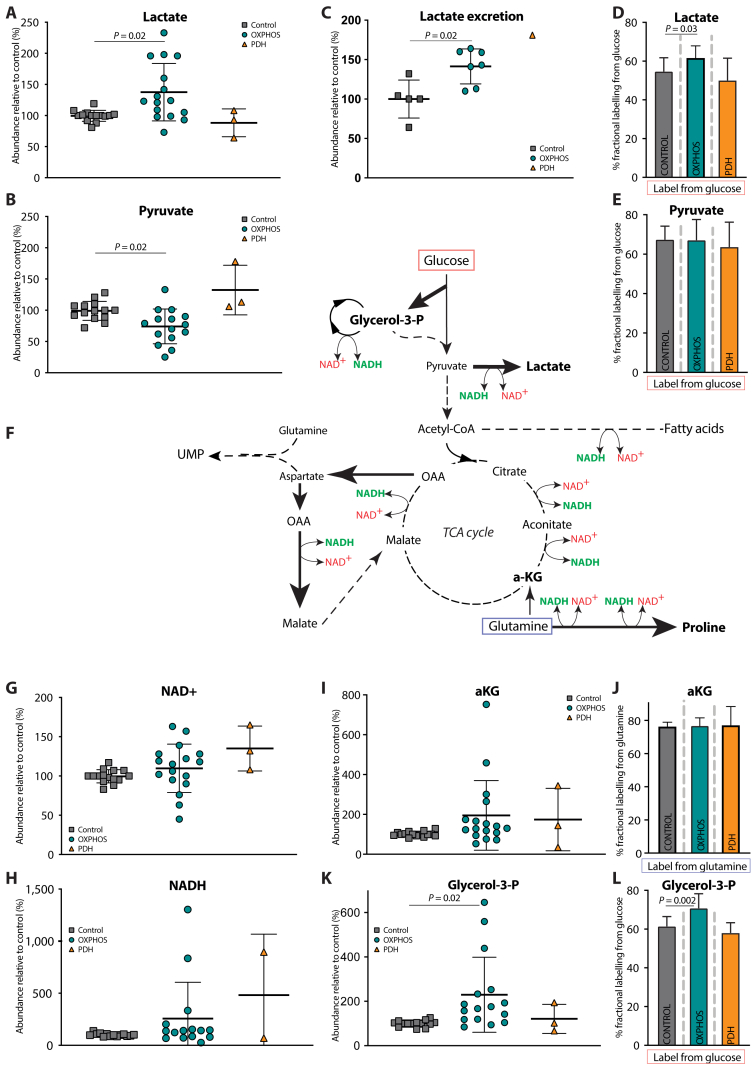
Results: OXPHOS dysfunction has a unique metabolic signature linked to an NAD+/NADH imbalance including depletion of TCA intermediates and aspartate, and increased levels of glycerol-3-phosphate. Supplementation with pyruvate and uridine fully rescues this altered metabolic profile and the subsequent proliferation deficit. Additionally, in zebrafish, the same nutritional treatment increases the survival after rotenone exposure.
Conclusions: Our findings reinforce the importance of the NAD+/NADH imbalance following OXPHOS dysfunction in PMD and open the door to new diagnostic and therapeutic tools for PMD.
Keywords: Aspartic acid; OXPHOS; Primary mitochondrial disease; Pyruvate and uridine; Treatment.
Copyright © 2022 The Author(s). Published by Elsevier GmbH.. All rights reserved.
Figures
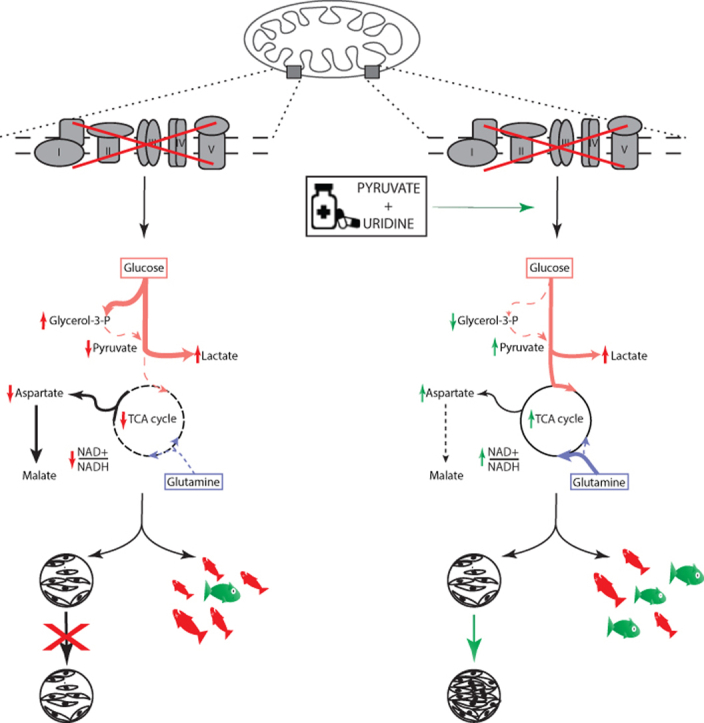
 OXPHOS deficiency;
OXPHOS deficiency;  PDH deficiency and
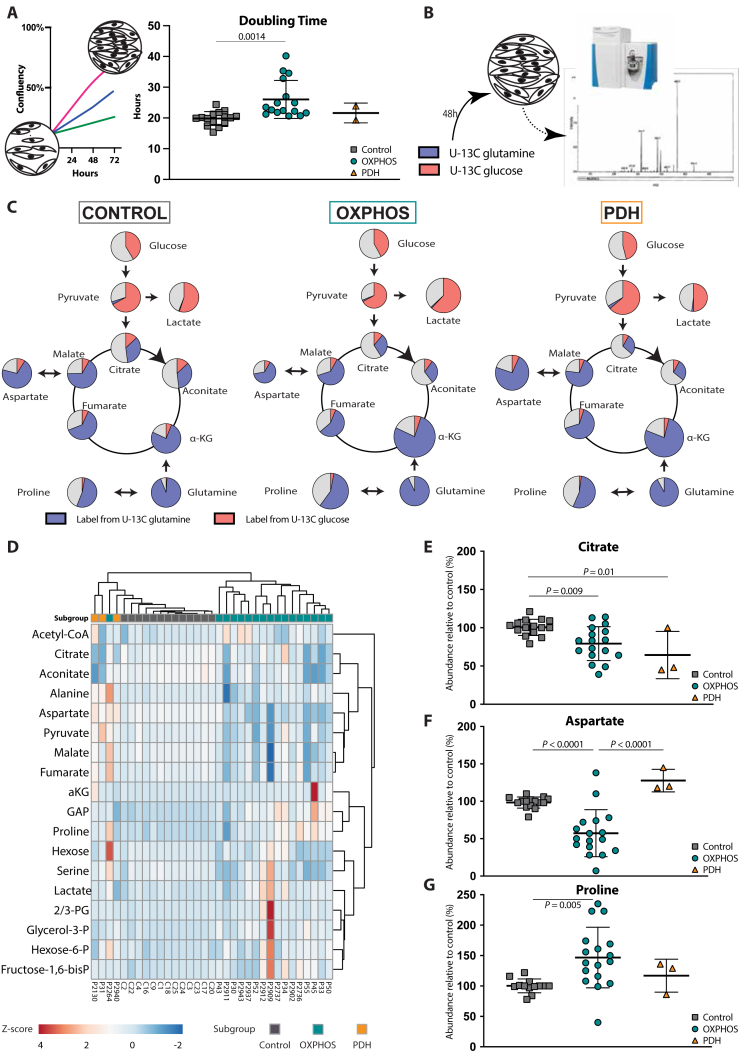
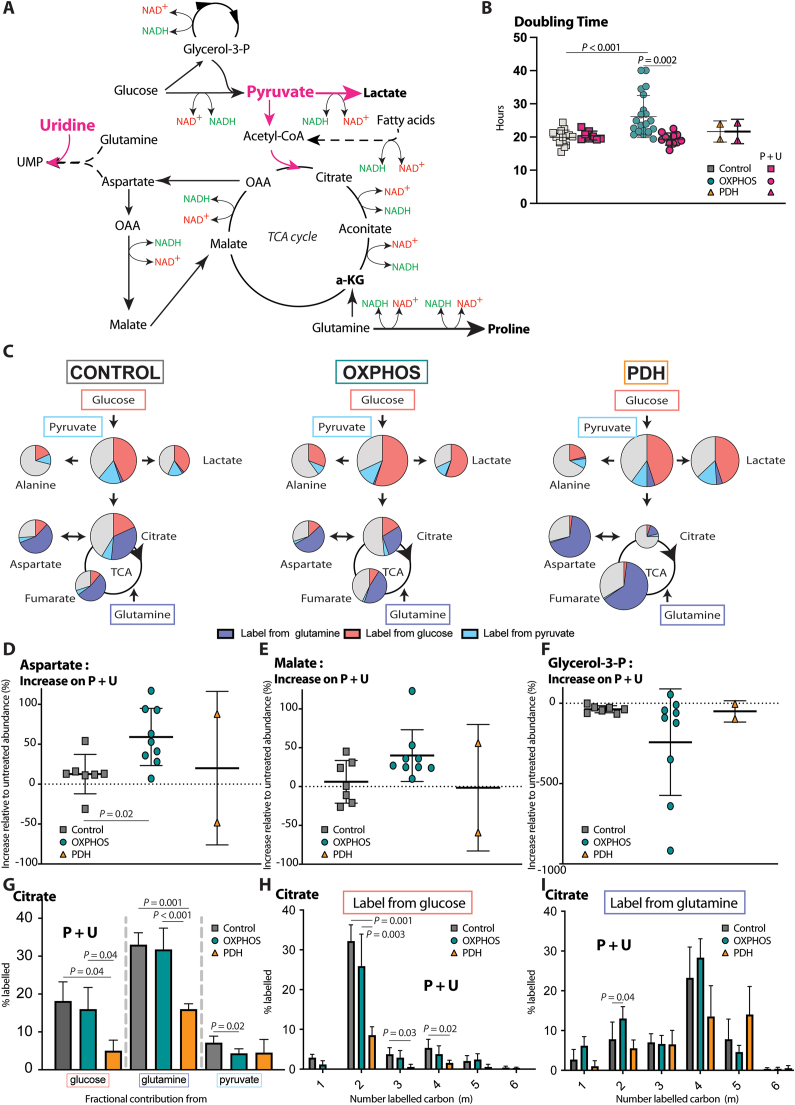
PDH deficiency and  controls. Full description of the genetic deficiency per cell line is described in Table 1. E The relative abundances of citrate, F aspartate and G proline in control, OXPHOS deficient and PDH deficient fibroblasts. Tracer metabolomics in control (n = 15), OXPHOS deficient (n = 17) and PDH deficient (n = 3) fibroblast cell lines (technical replicates 1–3 per cell line). Abundances normalized for protein content (BCA) and to the average of the controls per experiment. Statistics: one-way ANOVA with post-hoc Dunnett's T3 multiple comparison tests, and the error bars are +/−SD. αKG: alpha-ketoglutarate; CoA: coenzyme A; DHAP: dihydroxyacetone phosphate; Fructose-1,6-bisP: fructose-1,6-bisphosphate; GAP: glyceraldehyde-3-phosphate; Glycerol-3-P: Glycerol-3-phosphate; Hexose-6-P: Hexose-6-phosphate; OXPHOS: oxidative phosphorylation system; PDH: pyruvate dehydrogenase; PEP: phosphoenolpyruvate; TCA: tricarboxylic acid; 2/3-PG: 3-phosphoglycerate.
controls. Full description of the genetic deficiency per cell line is described in Table 1. E The relative abundances of citrate, F aspartate and G proline in control, OXPHOS deficient and PDH deficient fibroblasts. Tracer metabolomics in control (n = 15), OXPHOS deficient (n = 17) and PDH deficient (n = 3) fibroblast cell lines (technical replicates 1–3 per cell line). Abundances normalized for protein content (BCA) and to the average of the controls per experiment. Statistics: one-way ANOVA with post-hoc Dunnett's T3 multiple comparison tests, and the error bars are +/−SD. αKG: alpha-ketoglutarate; CoA: coenzyme A; DHAP: dihydroxyacetone phosphate; Fructose-1,6-bisP: fructose-1,6-bisphosphate; GAP: glyceraldehyde-3-phosphate; Glycerol-3-P: Glycerol-3-phosphate; Hexose-6-P: Hexose-6-phosphate; OXPHOS: oxidative phosphorylation system; PDH: pyruvate dehydrogenase; PEP: phosphoenolpyruvate; TCA: tricarboxylic acid; 2/3-PG: 3-phosphoglycerate.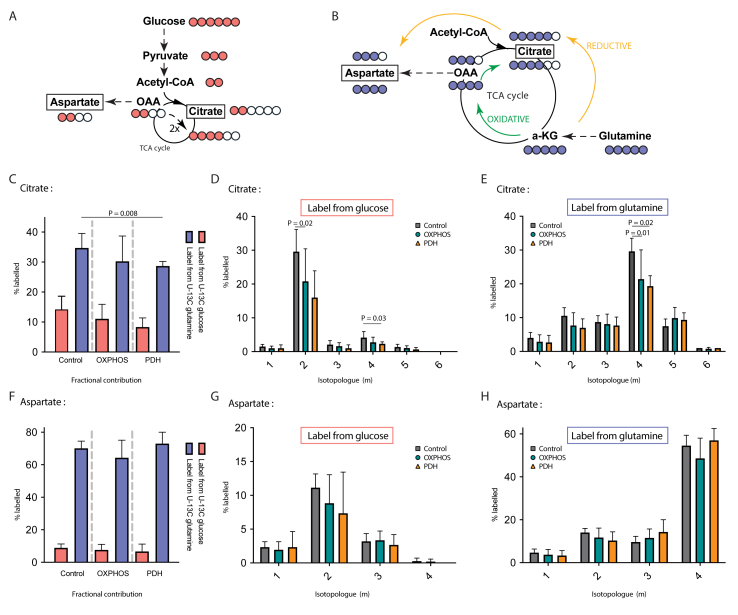
References
-
- Gorman G.S., Chinnery P.F., DiMauro S., Hirano M., Koga Y., McFarland R., et al. Mitochondrial diseases. Nature Reviews Disease Primers. 2016;2:1–23. - PubMed
-
- Distelmaier F., Koopman W.J.H., Van Den Heuvel L.P., Rodenburg R.J., Mayatepek E., Willems P.H.G.M., et al. Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain. 2009;132:833–842. - PubMed
-
- Hirst J. Mitochondrial complex i. Annual Review of Biochemistry. 2013;82:551–575. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
