

Reconstruction of a catalogue of genome-scale metabolic models with enzymatic constraints using GECKO 2.0
- PMID: 35773252
- PMCID: PMC9246944
- DOI: 10.1038/s41467-022-31421-1
Reconstruction of a catalogue of genome-scale metabolic models with enzymatic constraints using GECKO 2.0
Abstract
Genome-scale metabolic models (GEMs) have been widely used for quantitative exploration of the relation between genotype and phenotype. Streamlined integration of enzyme constraints and proteomics data into such models was first enabled by the GECKO toolbox, allowing the study of phenotypes constrained by protein limitations. Here, we upgrade the toolbox in order to enhance models with enzyme and proteomics constraints for any organism with a compatible GEM reconstruction. With this, enzyme-constrained models for the budding yeasts Saccharomyces cerevisiae, Yarrowia lipolytica and Kluyveromyces marxianus are generated to study their long-term adaptation to several stress factors by incorporation of proteomics data. Predictions reveal that upregulation and high saturation of enzymes in amino acid metabolism are common across organisms and conditions, suggesting the relevance of metabolic robustness in contrast to optimal protein utilization as a cellular objective for microbial growth under stress and nutrient-limited conditions. The functionality of GECKO is expanded with an automated framework for continuous and version-controlled update of enzyme-constrained GEMs, also producing such models for Escherichia coli and Homo sapiens. In this work, we facilitate the utilization of enzyme-constrained GEMs in basic science, metabolic engineering and synthetic biology purposes.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
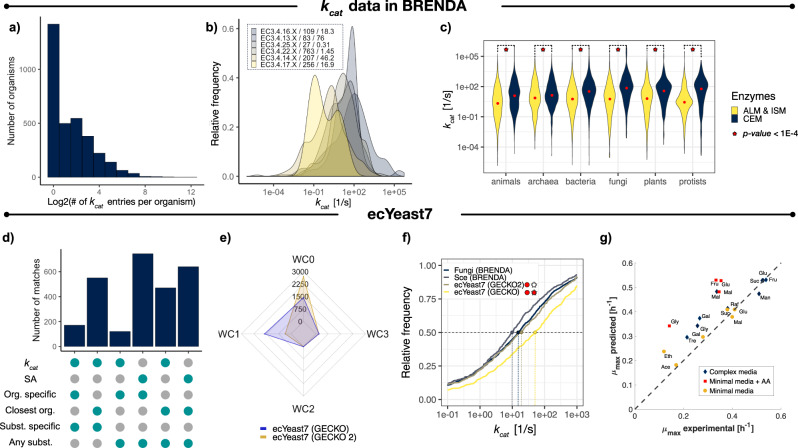
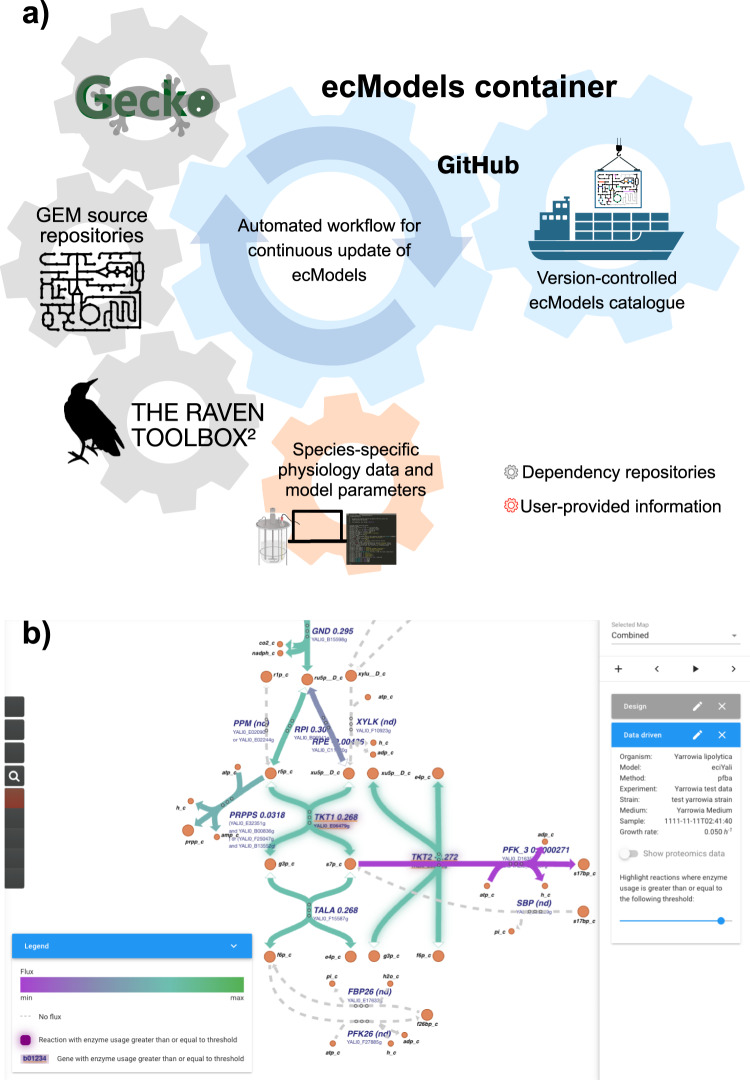
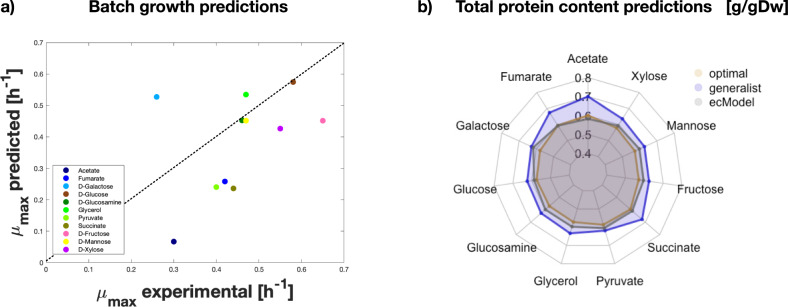
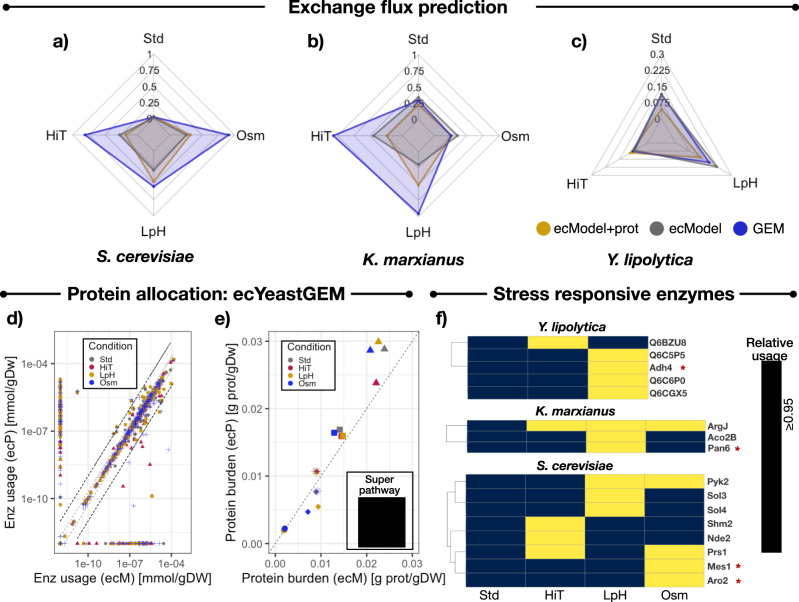

References
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
