

Dihydroorotate dehydrogenase inhibition reveals metabolic vulnerability in chronic myeloid leukemia
- PMID: 35773274
- PMCID: PMC9247109
- DOI: 10.1038/s41419-022-05028-9
Dihydroorotate dehydrogenase inhibition reveals metabolic vulnerability in chronic myeloid leukemia
Abstract
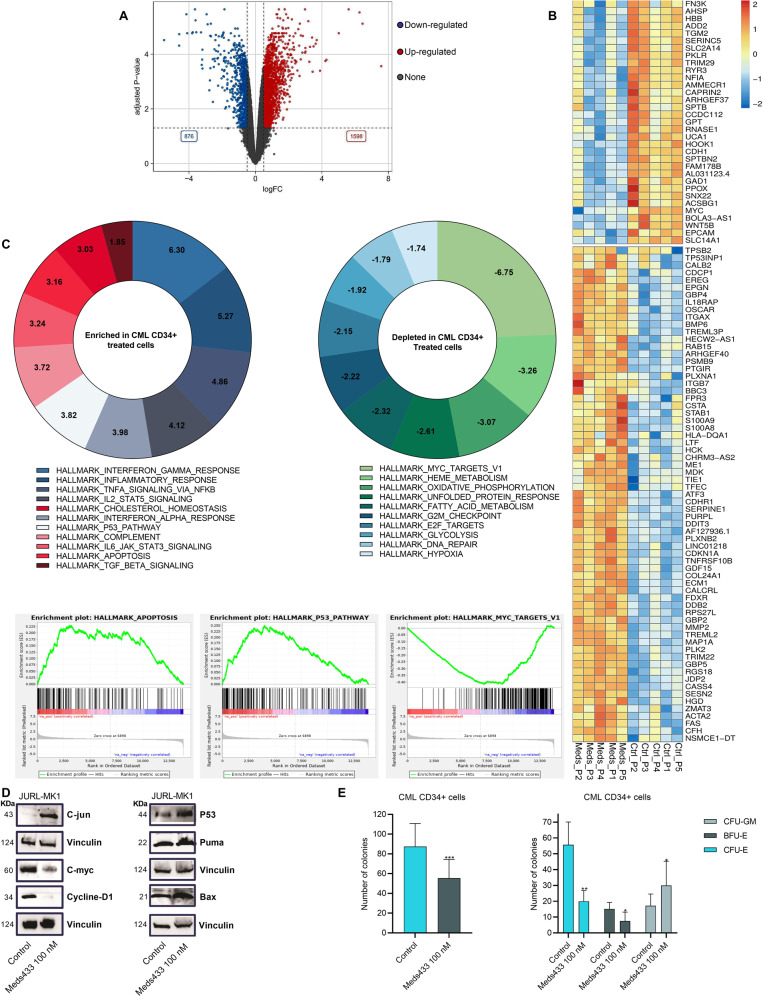
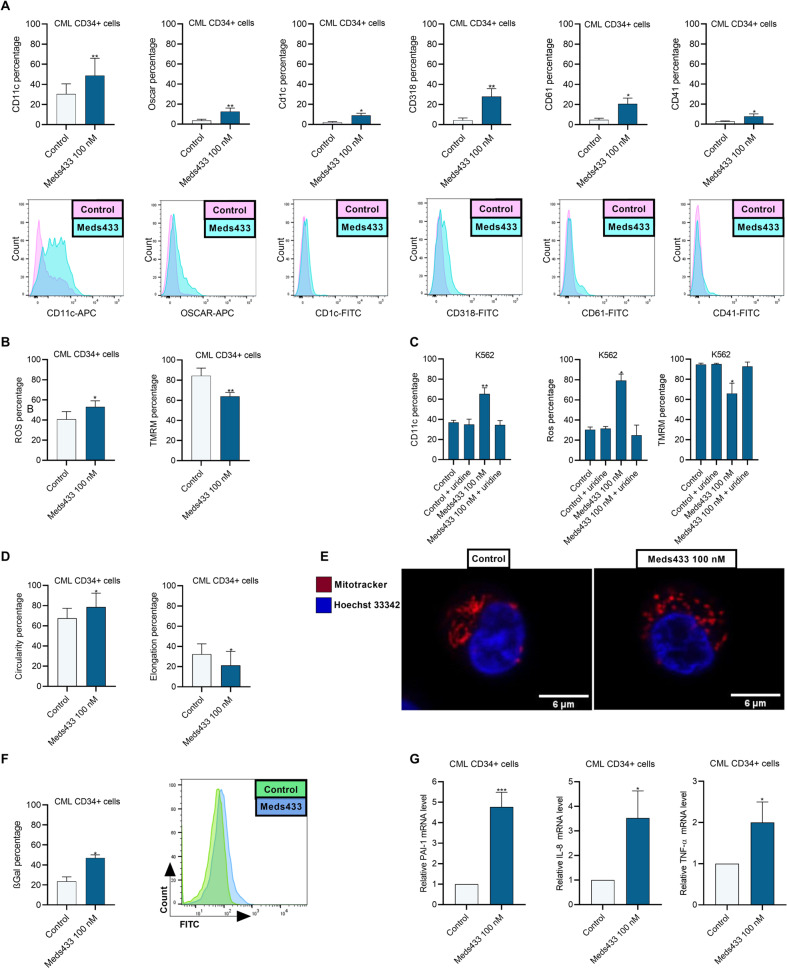
The development of different generations of BCR-ABL1 tyrosine kinase inhibitors (TKIs) has led to the high overall survival of chronic myeloid leukemia (CML) patients. However, there are CML patients who show resistance to TKI therapy and are prone to progress to more advanced phases of the disease. So, implementing an alternative approach for targeting TKIs insensitive cells would be of the essence. Dihydroorotate dehydrogenase (DHODH) is an enzyme in the de novo pyrimidine biosynthesis pathway that is located in the inner membrane of mitochondria. Here, we found that CML cells are vulnerable to DHODH inhibition mediated by Meds433, a new and potent DHODH inhibitor recently developed by our group. Meds433 significantly activates the apoptotic pathway and leads to the reduction of amino acids and induction of huge metabolic stress in CML CD34+ cells. Altogether, our study shows that DHODH inhibition is a promising approach for targeting CML stem/progenitor cells and may help more patients discontinue the therapy.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
