

Single-cell and bulk transcriptome sequencing identifies two epithelial tumor cell states and refines the consensus molecular classification of colorectal cancer
- PMID: 35773407
- PMCID: PMC9279158
- DOI: 10.1038/s41588-022-01100-4
Single-cell and bulk transcriptome sequencing identifies two epithelial tumor cell states and refines the consensus molecular classification of colorectal cancer
Abstract
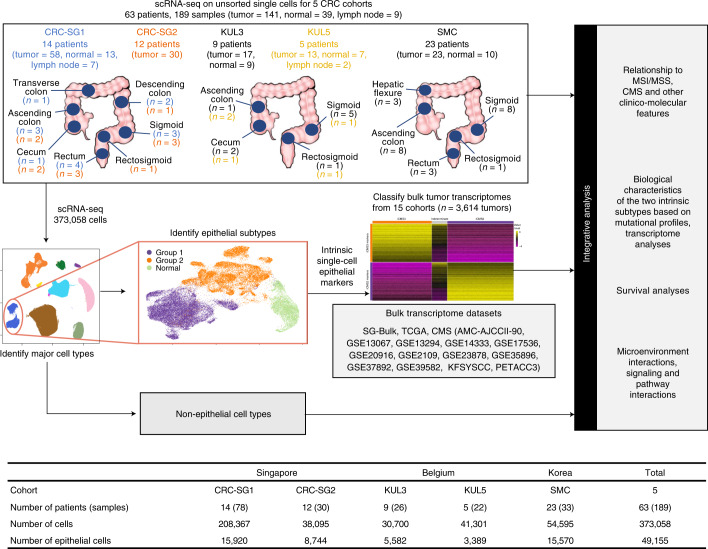
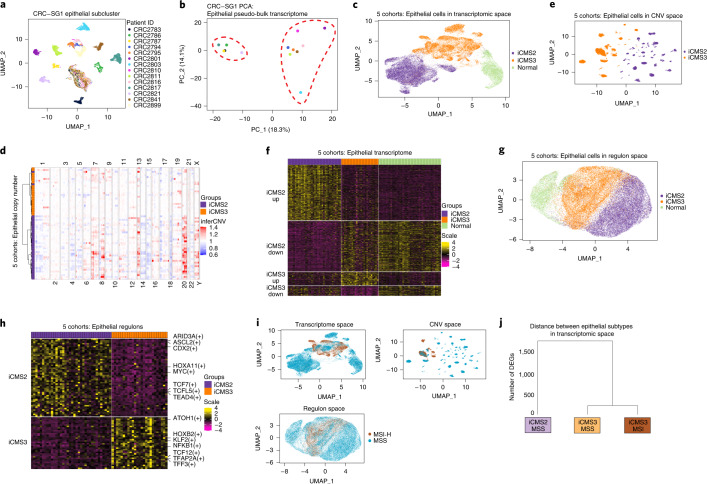
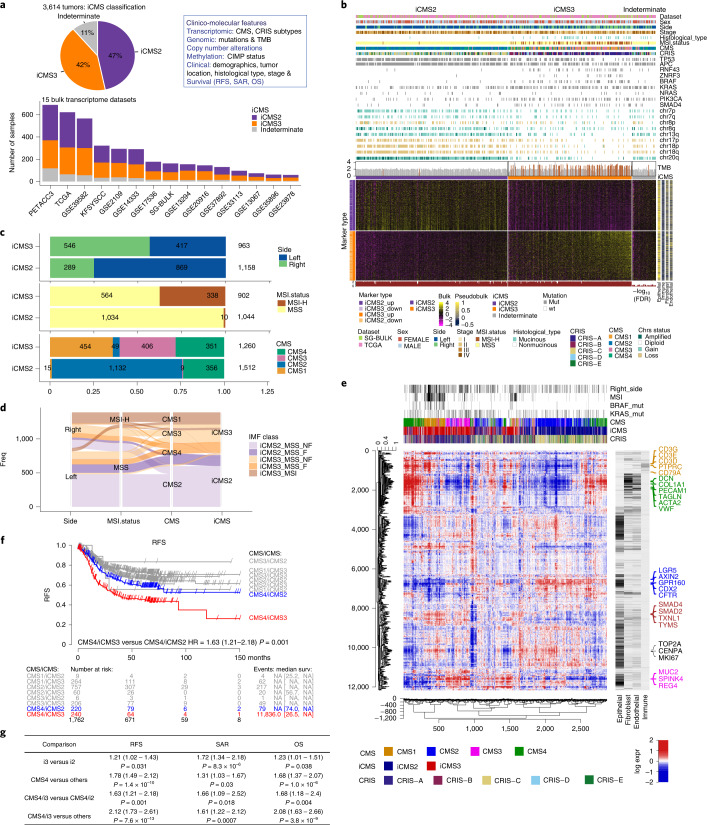
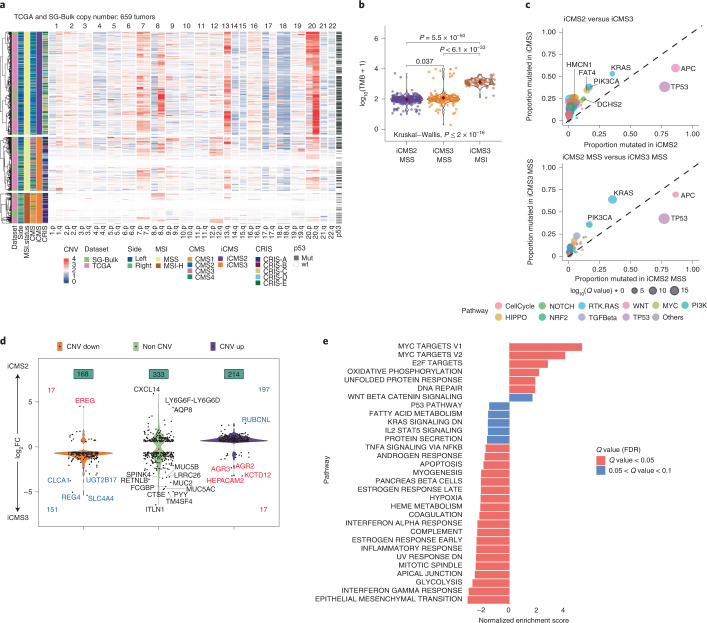
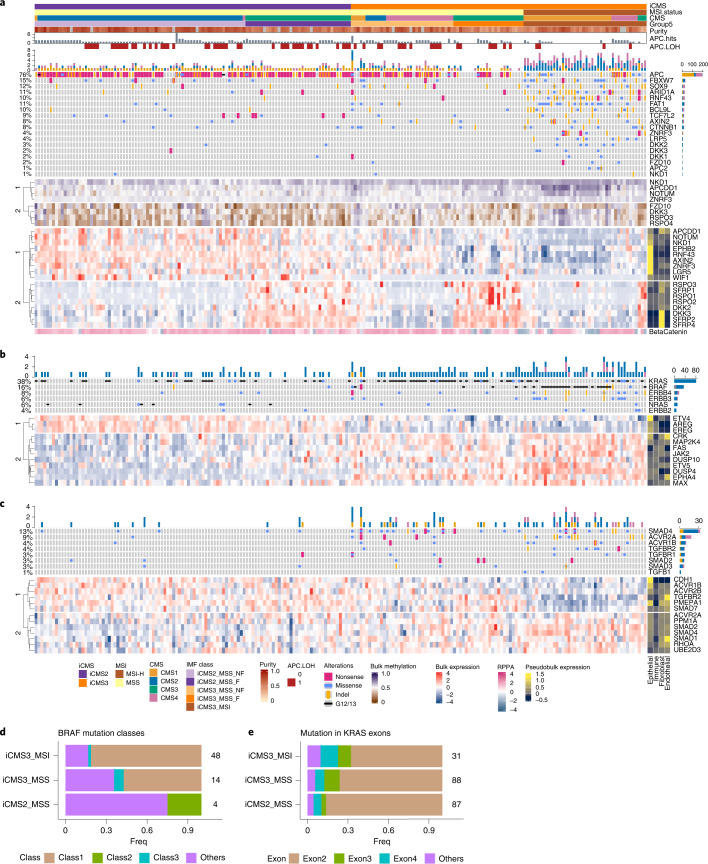
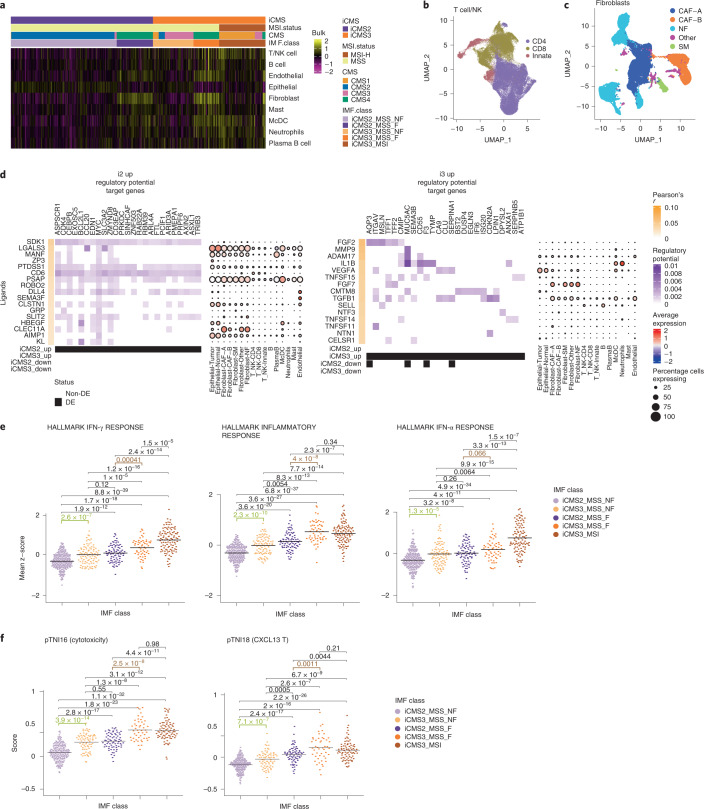
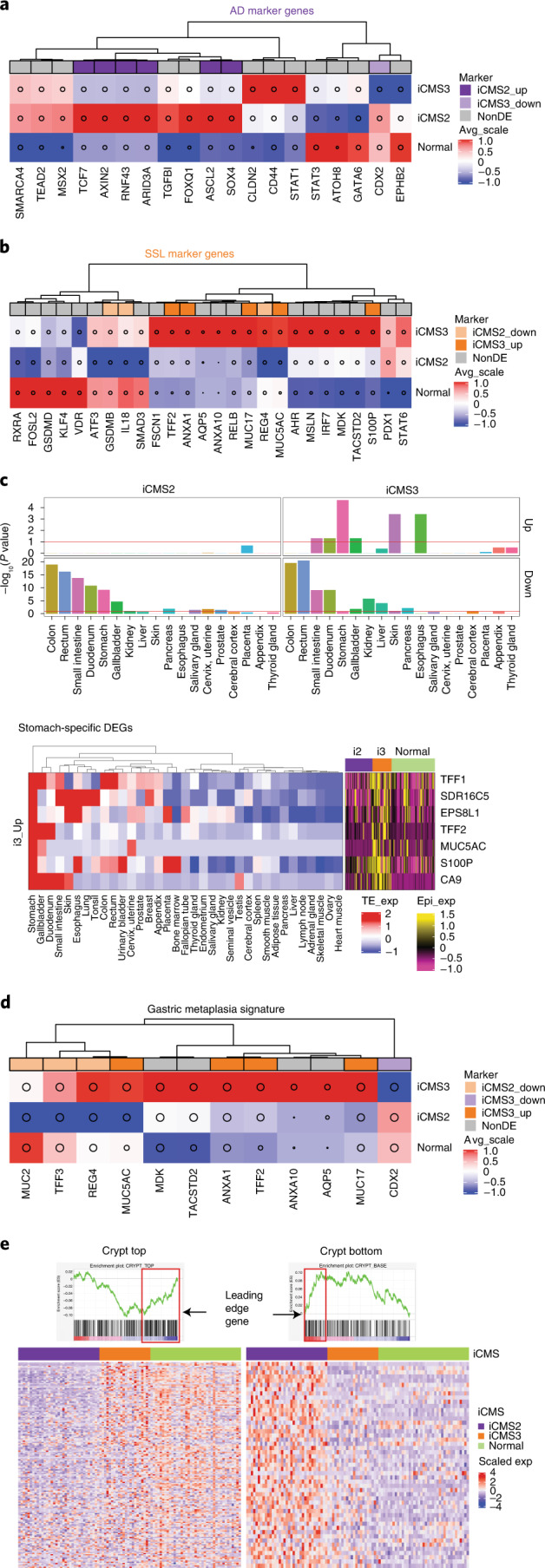
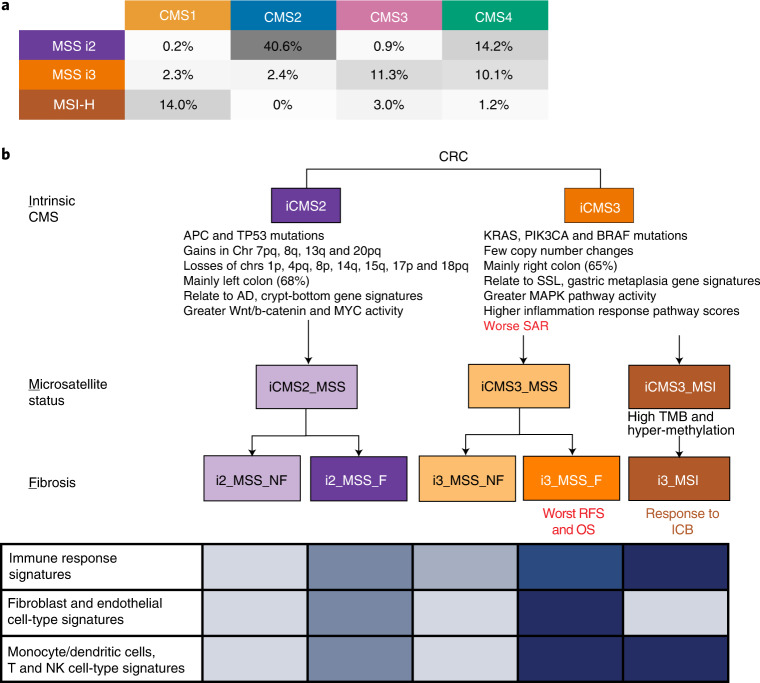
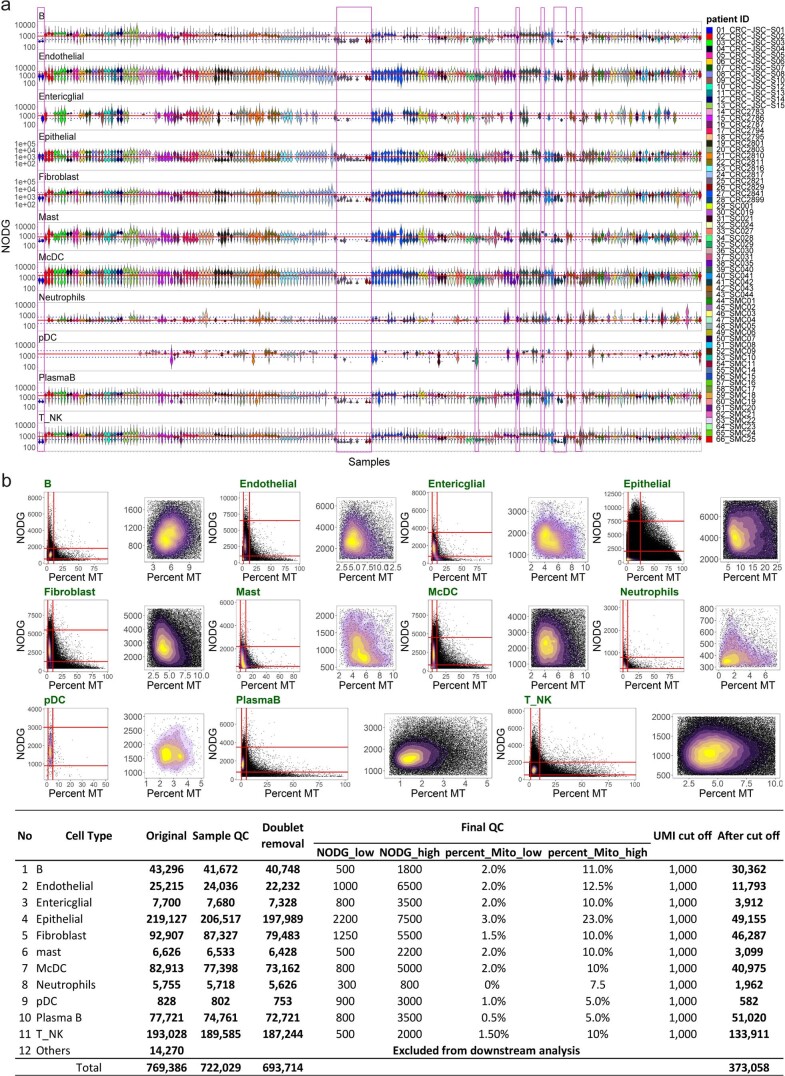
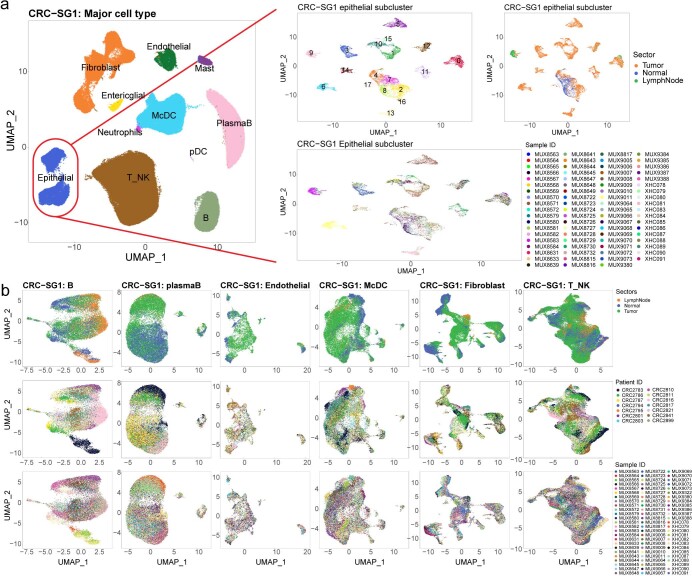
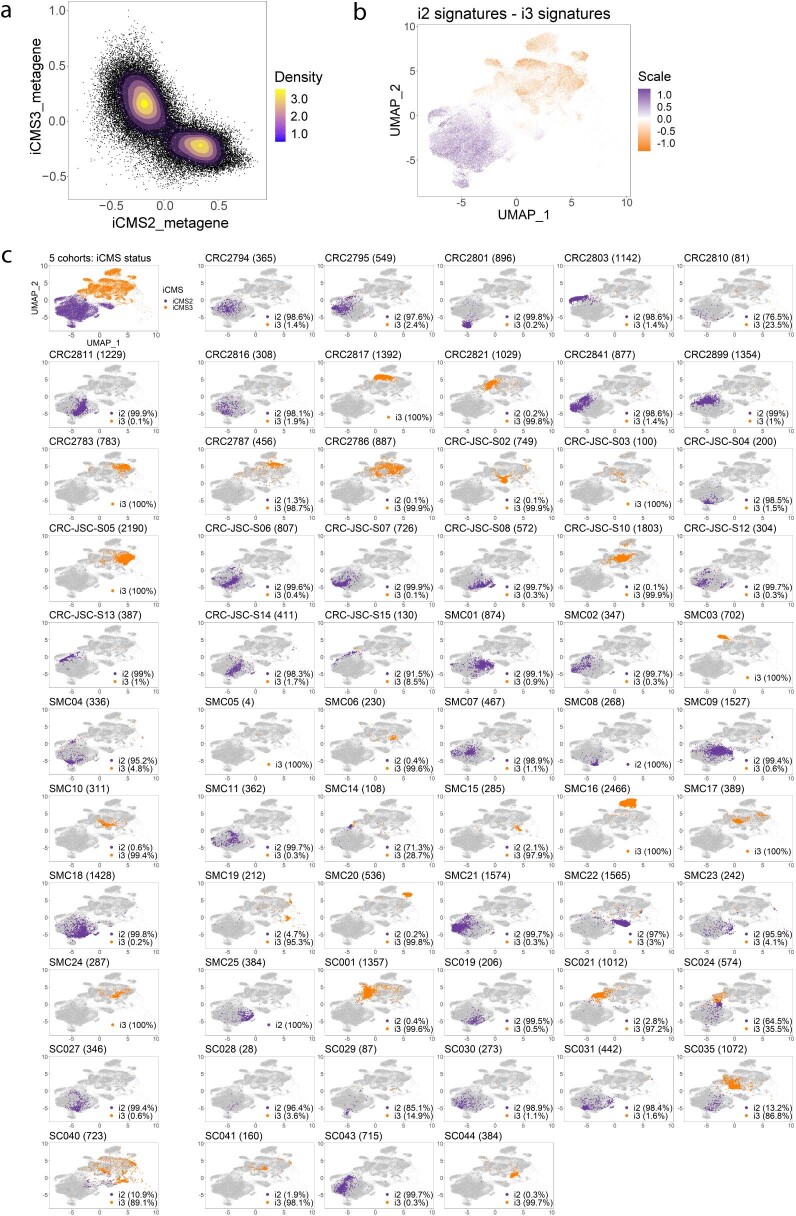
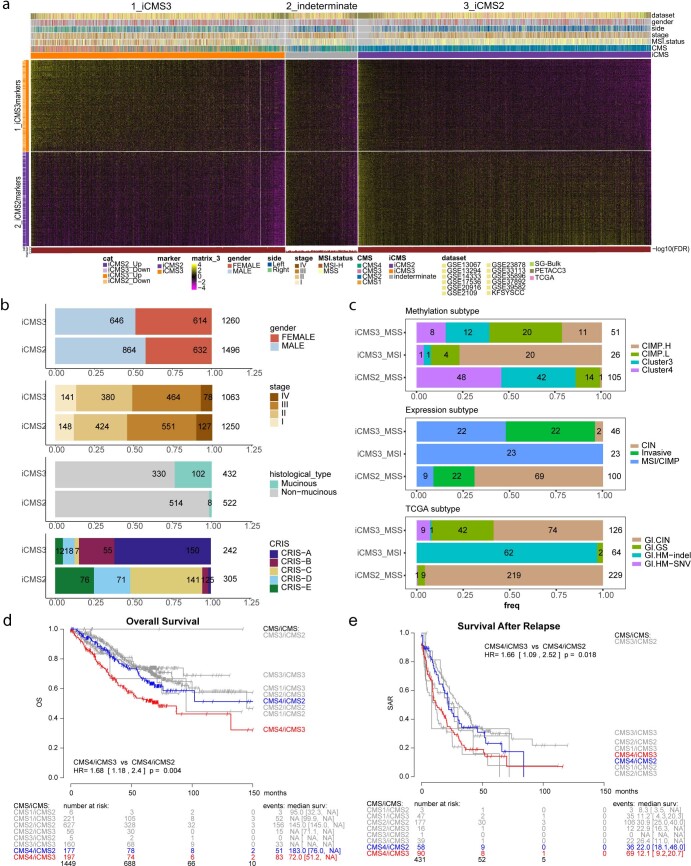
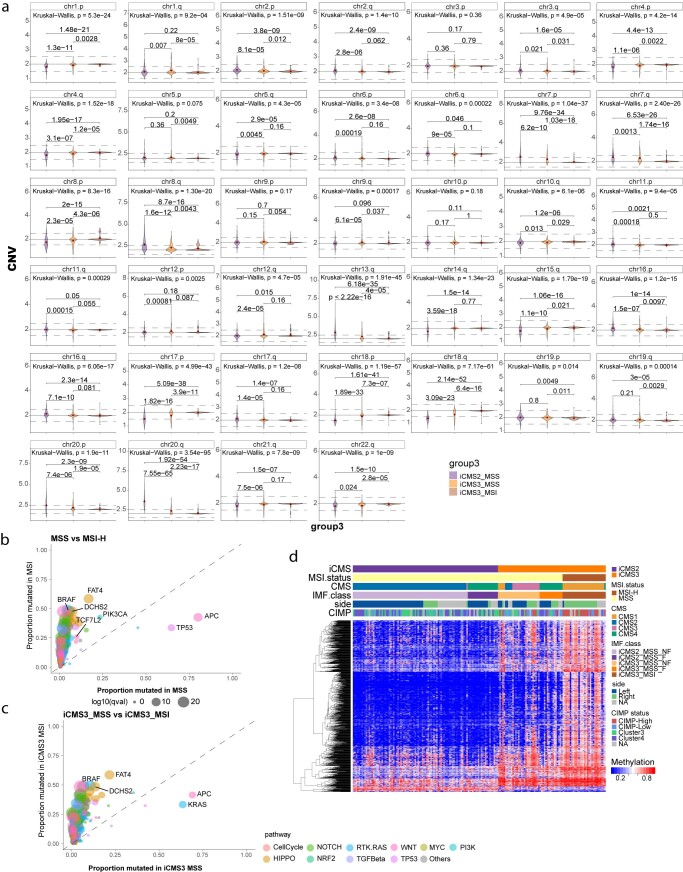
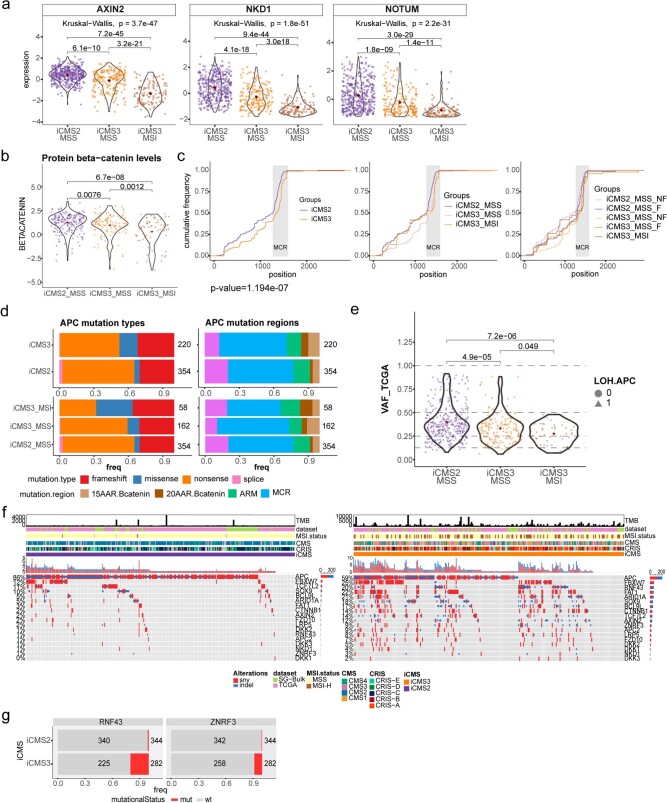
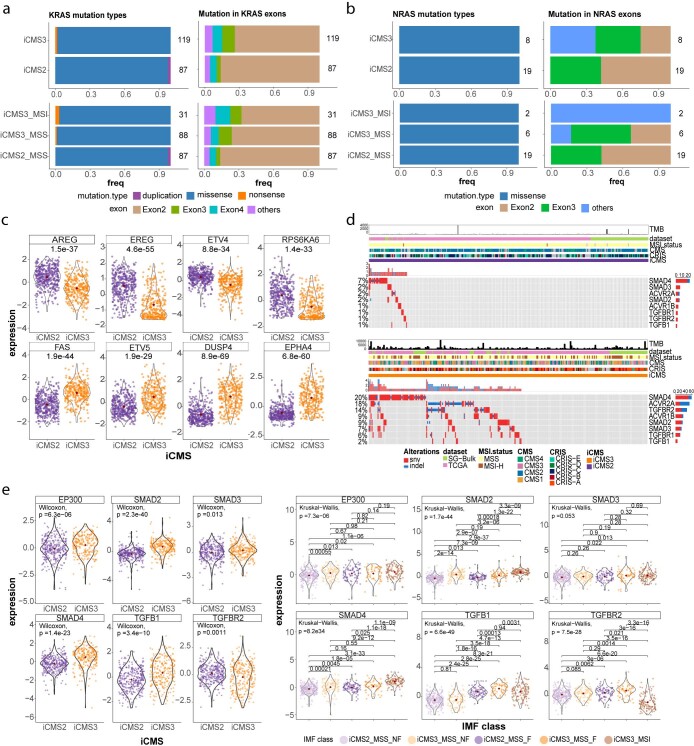
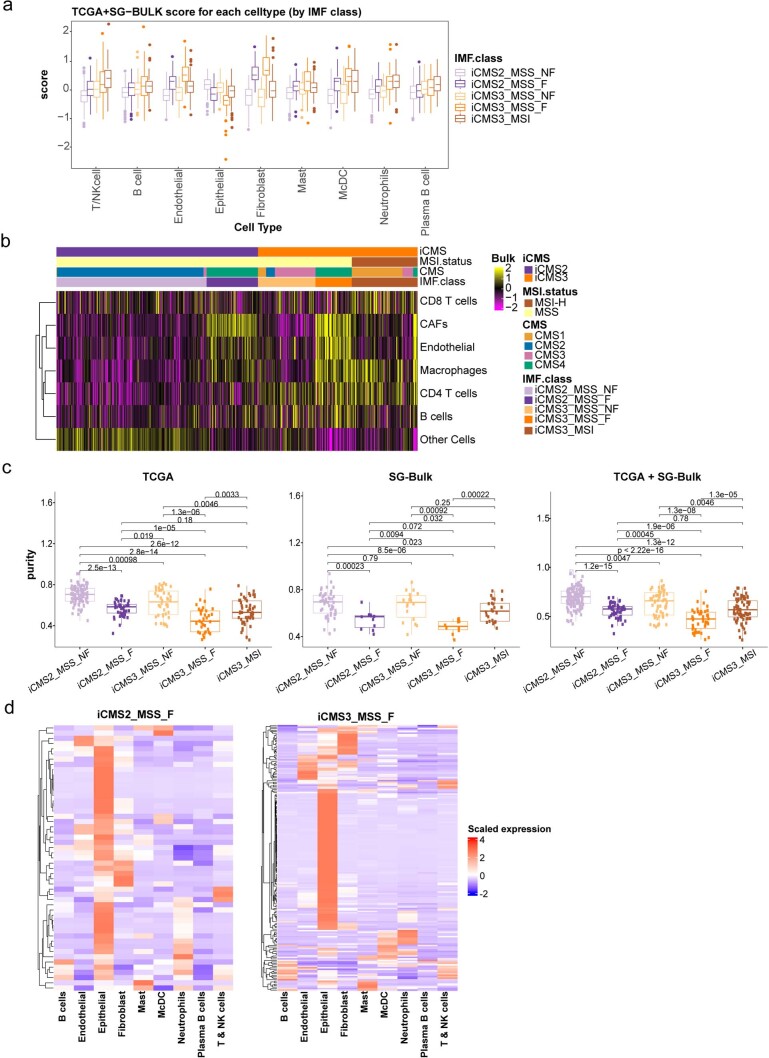
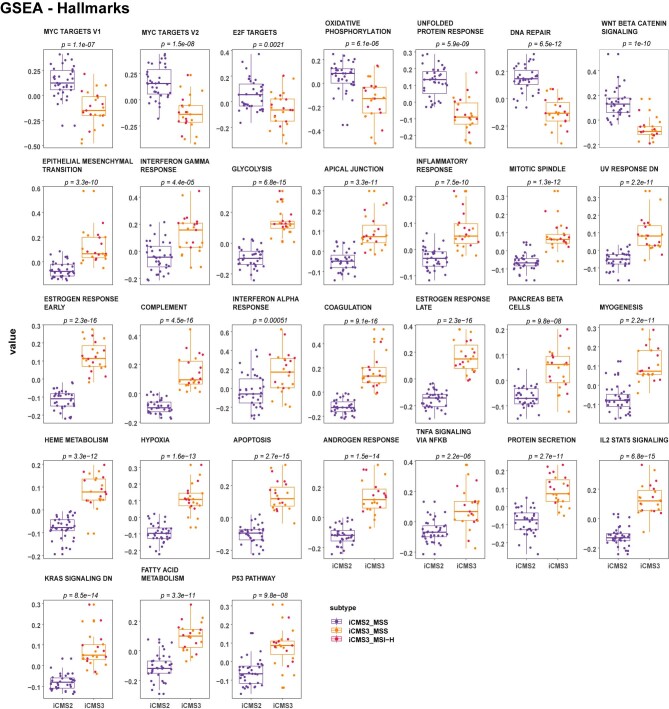
The consensus molecular subtype (CMS) classification of colorectal cancer is based on bulk transcriptomics. The underlying epithelial cell diversity remains unclear. We analyzed 373,058 single-cell transcriptomes from 63 patients, focusing on 49,155 epithelial cells. We identified a pervasive genetic and transcriptomic dichotomy of malignant cells, based on distinct gene expression, DNA copy number and gene regulatory network. We recapitulated these subtypes in bulk transcriptomes from 3,614 patients. The two intrinsic subtypes, iCMS2 and iCMS3, refine CMS. iCMS3 comprises microsatellite unstable (MSI-H) cancers and one-third of microsatellite-stable (MSS) tumors. iCMS3 MSS cancers are transcriptomically more similar to MSI-H cancers than to other MSS cancers. CMS4 cancers had either iCMS2 or iCMS3 epithelium; the latter had the worst prognosis. We defined the intrinsic epithelial axis of colorectal cancer and propose a refined 'IMF' classification with five subtypes, combining intrinsic epithelial subtype (I), microsatellite instability status (M) and fibrosis (F).
© 2022. The Author(s).
Conflict of interest statement
L.Z.H., R.N., H.L.J.O. and M.T.W. are employees of MSD International GmbH (Singapore Branch). A.N. and J.G. are employees of NantOmics. The remaining authors declare no competing interests.
Figures

Comment in
-
Hidden epithelial subtypes refine colorectal cancer classification.Nat Rev Cancer. 2022 Sep;22(9):495. doi: 10.1038/s41568-022-00501-1. Nat Rev Cancer. 2022. PMID: 35918514 No abstract available.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
