

PKD2 founder mutation is the most common mutation of polycystic kidney disease in Taiwan
- PMID: 35778421
- PMCID: PMC9249874
- DOI: 10.1038/s41525-022-00309-w
PKD2 founder mutation is the most common mutation of polycystic kidney disease in Taiwan
Abstract
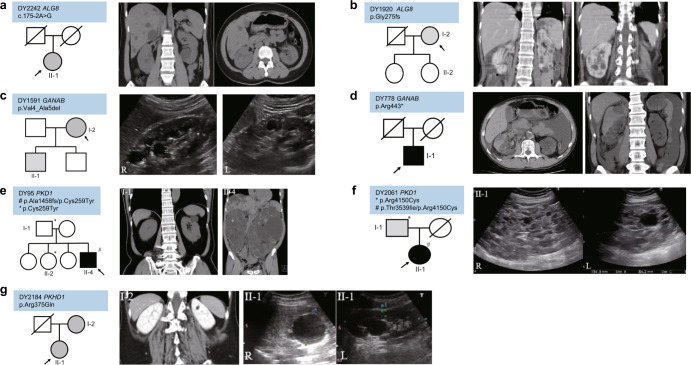
Autosomal Dominant polycystic kidney disease (ADPKD) is the most common inherited adult kidney disease. Although ADPKD is primarily caused by PKD1 and PKD2, the identification of several novel causative genes in recent years has revealed more complex genetic heterogeneity than previously thought. To study the disease-causing mutations of ADPKD, a total of 920 families were collected and their diagnoses were established via clinical and image studies by Taiwan PKD Consortium investigators. Amplicon-based library preparation with next-generation sequencing, variant calling, and bioinformatic analysis was used to identify disease-causing mutations in the cohort. Microsatellite analysis along with genotyping and haplotype analysis was performed in the PKD2 p.Arg803* family members. The age of mutation was calculated to estimate the time at which the mutation occurred or the founder arrived in Taiwan. Disease-causing mutations were identified in 634 families (68.9%) by detection of 364 PKD1, 239 PKD2, 18 PKHD1, 7 GANAB, and 6 ALG8 pathogenic variants. 162 families (17.6%) had likely causative but non-diagnostic variants of unknown significance (VUS). A single PKD2 p.Arg803* mutation was found in 17.8% (164/920) of the cohort in Taiwan. Microsatellite and array analysis showed that 80% of the PKD2 p.Arg803* families shared the same haplotype in a 250 kb region, indicating those families may originate from a common ancestor 300 years ago. Our findings provide a mutation landscape as well as evidence that a founder effect exists and has contributed to a major percentage of the ADPKD population in Taiwan.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Grants and funding
- CA-110-PP-08/National Health Research Institutes (NHRI)
- 104-2314-B-037-066/Ministry of Science and Technology, Taiwan (Ministry of Science and Technology of Taiwan)
- 108-2314-B-400-041-MY3/Ministry of Science and Technology, Taiwan (Ministry of Science and Technology of Taiwan)
- R01 DK068306/DK/NIDDK NIH HHS/United States
- 107-2314-B-400-041/Ministry of Science and Technology, Taiwan (Ministry of Science and Technology of Taiwan)
LinkOut - more resources
Full Text Sources
Miscellaneous
