

Metagenomic Analyses of Multiple Gut Datasets Revealed the Association of Phage Signatures in Colorectal Cancer
- PMID: 35782128
- PMCID: PMC9240273
- DOI: 10.3389/fcimb.2022.918010
Metagenomic Analyses of Multiple Gut Datasets Revealed the Association of Phage Signatures in Colorectal Cancer
Abstract
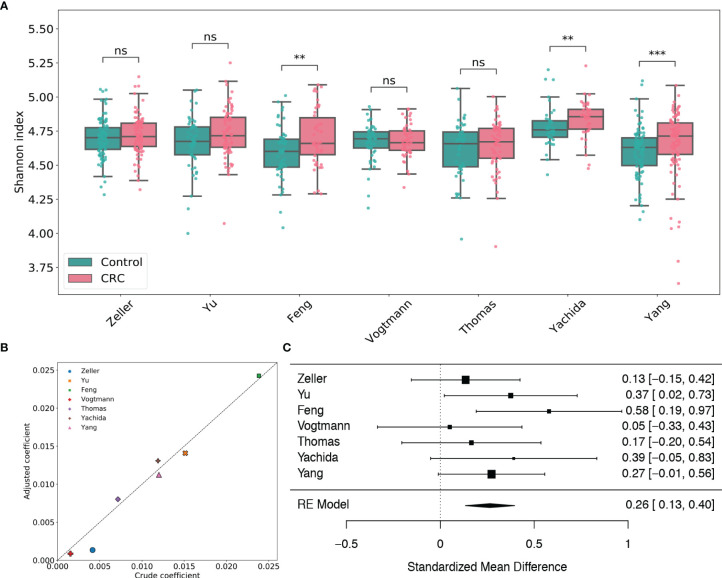
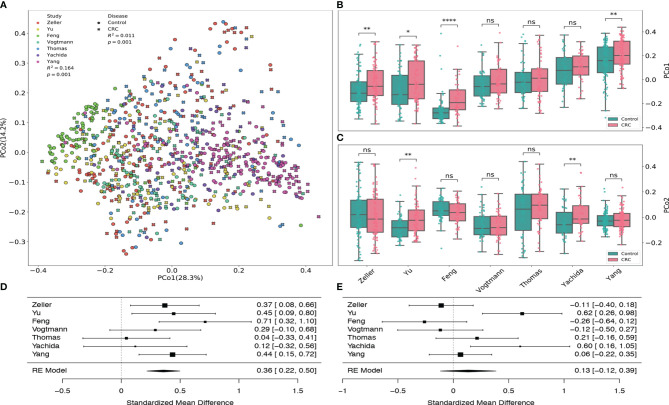
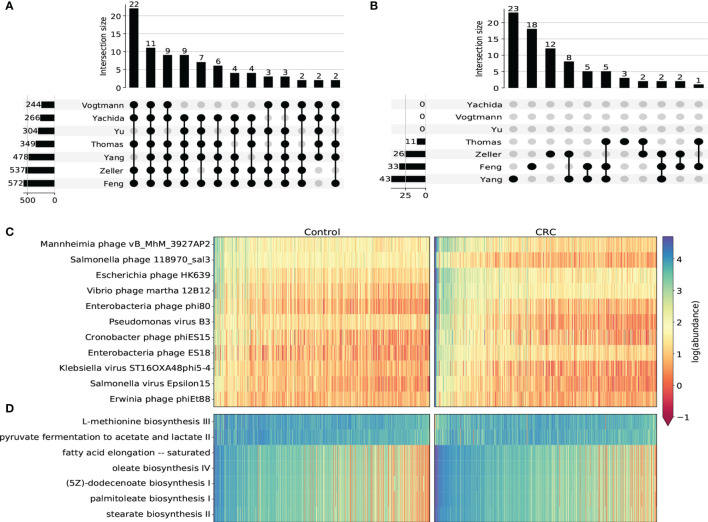
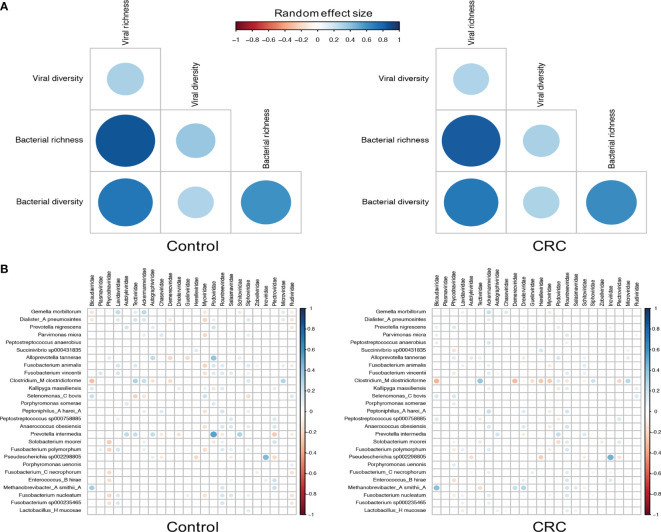
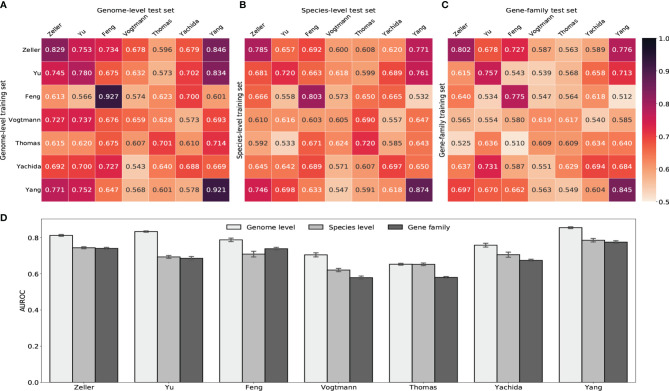
The association of colorectal cancer (CRC) and the human gut microbiome dysbiosis has been the focus of several studies in the past. Many bacterial taxa have been shown to have differential abundance among CRC patients compared to healthy controls. However, the relationship between CRC and non-bacterial gut microbiome such as the gut virome is under-studied and not well understood. In this study we conducted a comprehensive analysis of the association of viral abundances with CRC using metagenomic shotgun sequencing data of 462 CRC subjects and 449 healthy controls from 7 studies performed in 8 different countries. Despite the high heterogeneity, our results showed that the virome alpha diversity was consistently higher in CRC patients than in healthy controls (p-value <0.001). This finding is in sharp contrast to previous reports of low alpha diversity of prokaryotes in CRC compared to healthy controls. In addition to the previously known association of Podoviridae, Siphoviridae and Myoviridae with CRC, we further demonstrate that Herelleviridae, a newly constructed viral family, is significantly depleted in CRC subjects. Our interkingdom association analysis reveals a less intertwined correlation between the gut virome and bacteriome in CRC compared to healthy controls. Furthermore, we show that the viral abundance profiles can be used to accurately predict CRC disease status (AUROC >0.8) in both within-study and cross-study settings. The combination of training sets resulted in rather generalized and accurate prediction models. Our study clearly shows that subjects with colorectal cancer harbor a distinct human gut virome profile which may have an important role in this disease.
Keywords: CRC prediction; colorectal cancer; gut virome; metagenomics; virus-host association.
Copyright © 2022 Zuo, Michail and Sun.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Anderson M. J. (2001). A New Method for Non-Parametric Multivariate Analysis of Variance. Austral Ecol. 26 (1), 32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x - DOI
-
- Benjamini Y., Hochberg Y. (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Society: Ser. B. (Methodol.) 57 (1), 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous