

A novel HADHA variant associated with an atypical moderate and late-onset LCHAD deficiency
- PMID: 35782617
- PMCID: PMC9248219
- DOI: 10.1016/j.ymgmr.2022.100860
A novel HADHA variant associated with an atypical moderate and late-onset LCHAD deficiency
Abstract
Background: Long chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD) is a rare inherited disease caused by pathogenic variants of HADHA gene. Along with signs common to fatty acid oxidation defects (FAOD), specific retina and heart alterations are observed. Because long-chain fatty acid oxidation is selectively affected, supplementations with short/medium-chain fats represent energetic sources bypassing the enzymatic blockade. Here, we report on an atypical presentation of the disease.
Methods: Clinical features were described with medical explorations including ophthalmic and cardiac examination. Biological underlying defects were investigated by measurements of biochemical metabolites and by fluxomic studies of mitochondrial β-oxidation. Whole exome sequencing and molecular validation of variants confirmed the diagnosis.
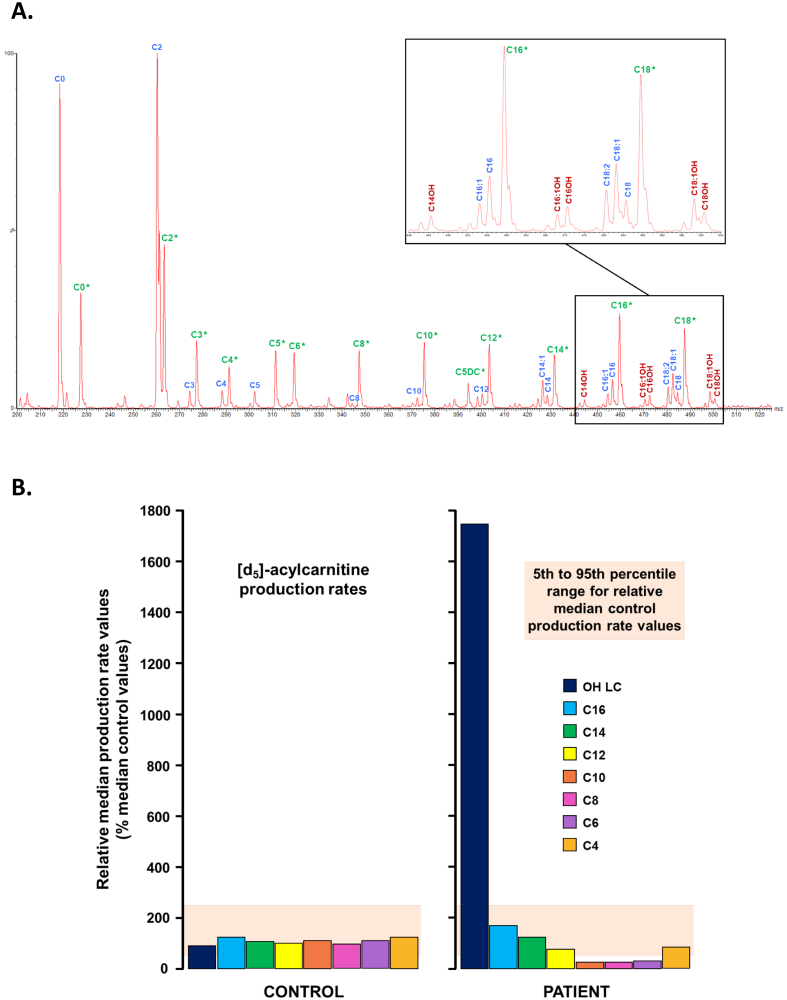
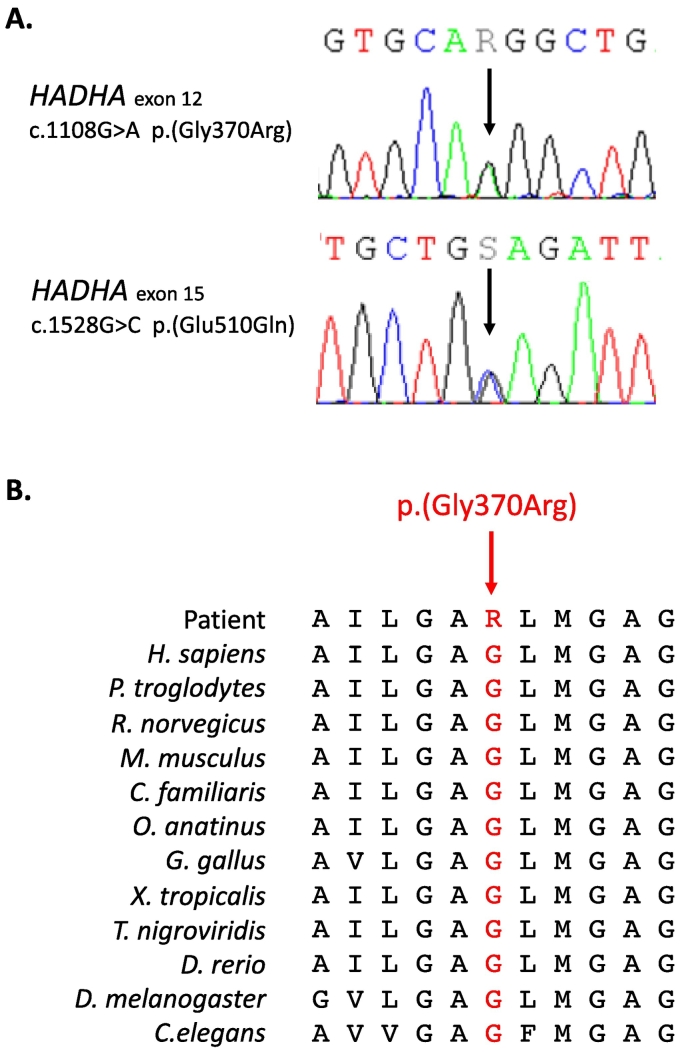
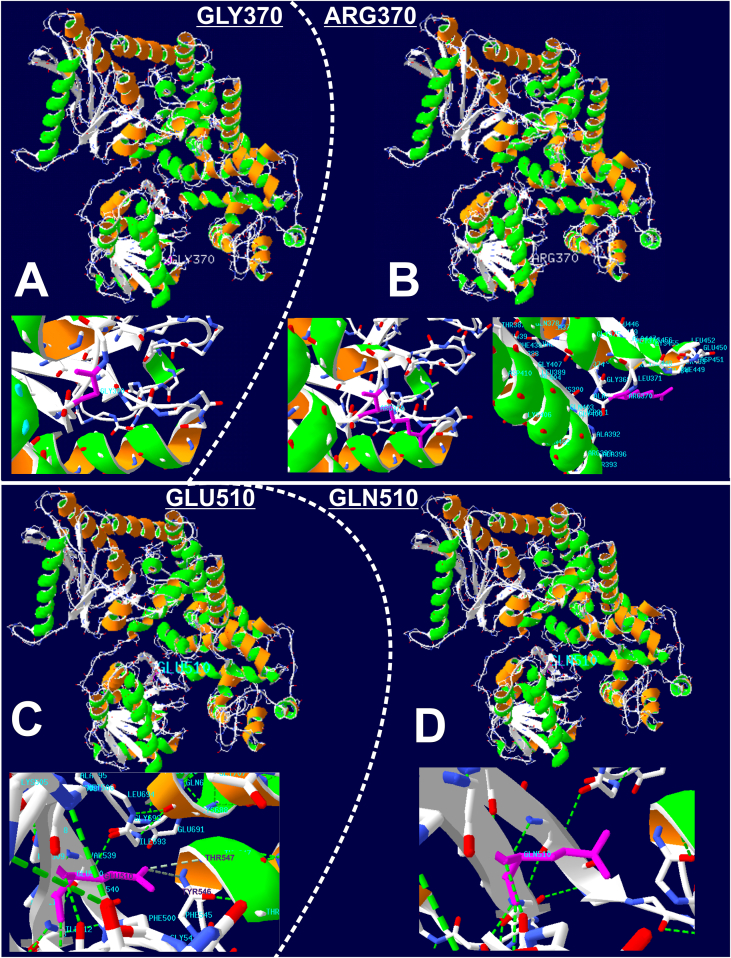
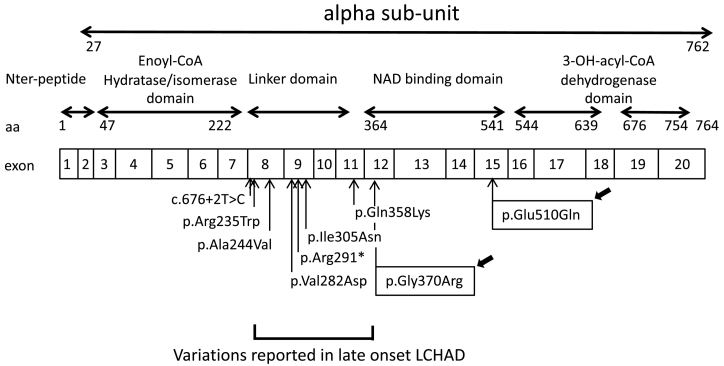
Results: The patient has developed at nine years an unlabeled maculopathy, and at 28 years, an acute cardiac decompensation without any premise. Blood individual acylcarnitine analysis showed a rise in hydroxylated long-chain fatty acids and fluxomic studies validated enzyme blockade consistent with LCHADD. Genetic analysis revealed the common p.(Glu510Gln) variant in HADHA, in trans with a novel variant c.1108G > A, p.(Gly370Arg) located in the NAD binding domain. Patient pathology was responsive to triheptanoin supplementation.
Conclusion: This atypical LCHADD form report should encourage the early assessment of biochemical and genetic testing as a specific management is recommended (combination with fast avoidance, low fat-high carbohydrate diet, medium-even-chain triglycerides or triheptanoin supplementation).
Keywords: Atypical maculopathy; Cardiomyopathy; HADHA; LCHAD; Late-onset; Mitochondrial trifunctional protein MTP.
© 2022 The Authors.
Conflict of interest statement
No conflicting relationship exists for any author.
Figures
References
-
- Boutron A., Acquaviva C., Vianey-Saban C., de Lonlay P., de Baulny H.O., Guffon N., Dobbelaere D., Feillet F., Labarthe F., Lamireau D., et al. Comprehensive CDNA study and quantitative analysis of mutant HADHA and HADHB transcripts in a french cohort of 52 patients with mitochondrial trifunctional protein deficiency. Mol. Genet. Metab. 2011;103(4):341–348. doi: 10.1016/j.ymgme.2011.04.006. - DOI - PubMed
-
- Lotz-Havla A.S., Röschinger W., Schiergens K., Singer K., Karall D., Konstantopoulou V., Wortmann S.B., Maier E.M. Fatal pitfalls in newborn screening for mitochondrial trifunctional protein (MTP)/Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. Orphanet J. Rare Dis. 2018;13(1):122. doi: 10.1186/s13023-018-0875-6. - DOI - PMC - PubMed
-
- Tyni T., Kivelä T., Lappi M., Summanen P., Nikoskelainen E., Pihko H. Ophthalmologic findings in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation: a new type of hereditary metabolic chorioretinopathy. Ophthalmology. 1998;105(5):810–824. doi: 10.1016/S0161-6420(98)95019-9. - DOI - PubMed
-
- De Biase I., Viau K.S., Liu A., Yuzyuk T., Botto L.D., Pasquali M., Longo N. Diagnosis, treatment, and clinical outcome of patients with mitochondrial trifunctional Protein/Long-chain 3-hydroxy acyl-CoA dehydrogenase deficiency. JIMD Rep. 2017;31:63–71. doi: 10.1007/8904_2016_558. - DOI - PMC - PubMed
-
- Tyni T., Pihko H., Kivelä T. Ophthalmic pathology in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation. Curr. Eye Res. 1998;17(6):551–559. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
