

High Diversity of Novel Viruses in the Tree Pathogen Phytophthora castaneae Revealed by High-Throughput Sequencing of Total and Small RNA
- PMID: 35783401
- PMCID: PMC9244493
- DOI: 10.3389/fmicb.2022.911474
High Diversity of Novel Viruses in the Tree Pathogen Phytophthora castaneae Revealed by High-Throughput Sequencing of Total and Small RNA
Abstract
Phytophthora castaneae, an oomycete pathogen causing root and trunk rot of different tree species in Asia, was shown to harbor a rich diversity of novel viruses from different families. Four P. castaneae isolates collected from Chamaecyparis hodginsii in a semi-natural montane forest site in Vietnam were investigated for viral presence by traditional and next-generation sequencing (NGS) techniques, i.e., double-stranded RNA (dsRNA) extraction and high-throughput sequencing (HTS) of small RNAs (sRNAs) and total RNA. Genome organization, sequence similarity, and phylogenetic analyses indicated that the viruses were related to members of the order Bunyavirales and families Endornaviridae, Megabirnaviridae, Narnaviridae, Totiviridae, and the proposed family "Fusagraviridae." The study describes six novel viruses: Phytophthora castaneae RNA virus 1-5 (PcaRV1-5) and Phytophthora castaneae negative-stranded RNA virus 1 (PcaNSRV1). All six viruses were detected by sRNA sequencing, which demonstrates an active RNA interference (RNAi) system targeting viruses in P. castaneae. To our knowledge, this is the first report of viruses in P. castaneae and the whole Phytophthora major Clade 5, as well as of the activity of an RNAi mechanism targeting viral genomes among Clade 5 species. PcaRV1 is the first megabirnavirus described in oomycetes and the genus Phytophthora.
Keywords: RNA interference; RdRp; dsRNA; forest pathogen; multiple viral infections; mycovirus; oomycetes; ssRNA.
Copyright © 2022 Raco, Vainio, Sutela, Eichmeier, Hakalová, Jung and Botella.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
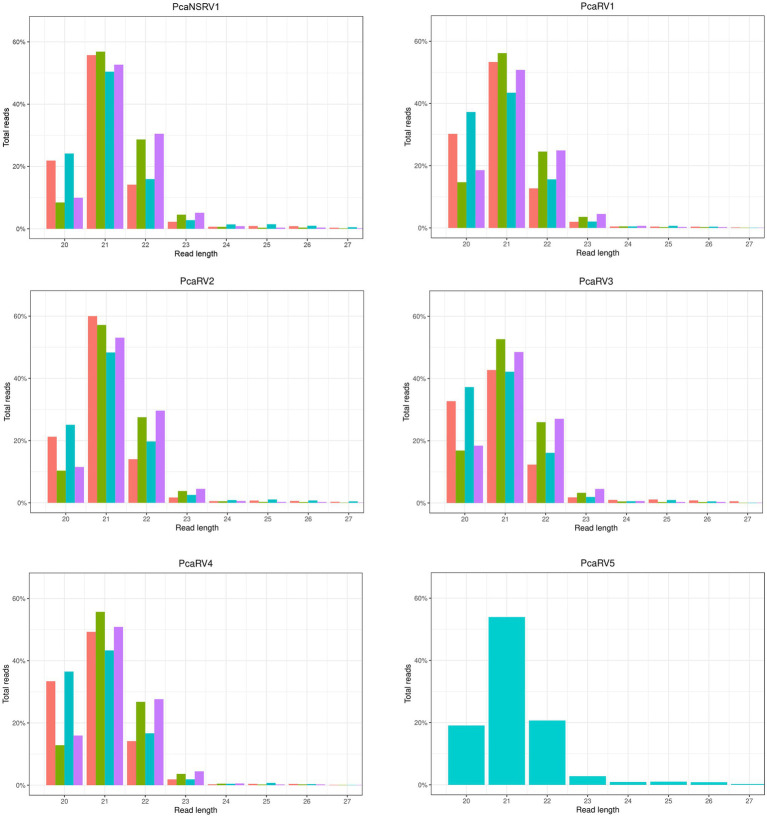
 ,VN1004
,VN1004  , VN1008
, VN1008  , and VN1012
, and VN1012  reads, respectively, after mapping to the final genomes of viruses PcaNSRV1 and PcaRV1-5, respectively. Mapping was done in Geneious Prime® 2020.2.3 using Bowtie mapper. The final graphs were constructed in R Core Team (2020). Sizes of sRNA profiles starting from 28 nt that were represented with less than 1.5% of total reads per virus were omitted during graph preparation for a better visualization.
reads, respectively, after mapping to the final genomes of viruses PcaNSRV1 and PcaRV1-5, respectively. Mapping was done in Geneious Prime® 2020.2.3 using Bowtie mapper. The final graphs were constructed in R Core Team (2020). Sizes of sRNA profiles starting from 28 nt that were represented with less than 1.5% of total reads per virus were omitted during graph preparation for a better visualization.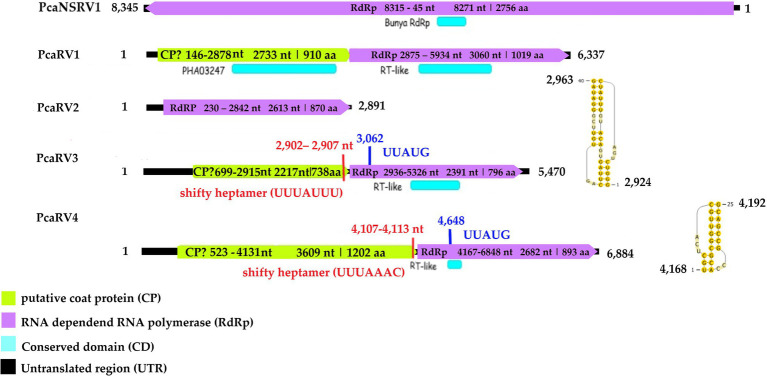
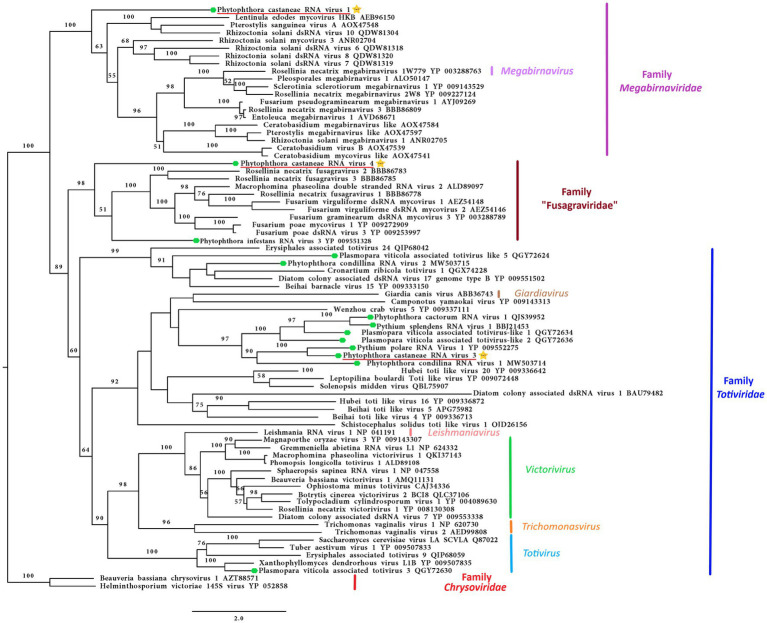
 ), and related members belonging to the families Megabirnaviridae, Toriviridae proposed family “Fusagraviridae.” The viruses described from oomycetes are indicated by a green hexagone
), and related members belonging to the families Megabirnaviridae, Toriviridae proposed family “Fusagraviridae.” The viruses described from oomycetes are indicated by a green hexagone  . Nodes are labeled with bootstrap percentages ≥50% only. Family classification and the corresponding pBLAST accession numbers are shown next to the virus names. The tree is rooted in the midpoint. Branch lengths are scaled to the expected underlying number of amino acid substitutions per site. The scale bar is indicatig 2.0 aa substitutions per site, per branch.
. Nodes are labeled with bootstrap percentages ≥50% only. Family classification and the corresponding pBLAST accession numbers are shown next to the virus names. The tree is rooted in the midpoint. Branch lengths are scaled to the expected underlying number of amino acid substitutions per site. The scale bar is indicatig 2.0 aa substitutions per site, per branch.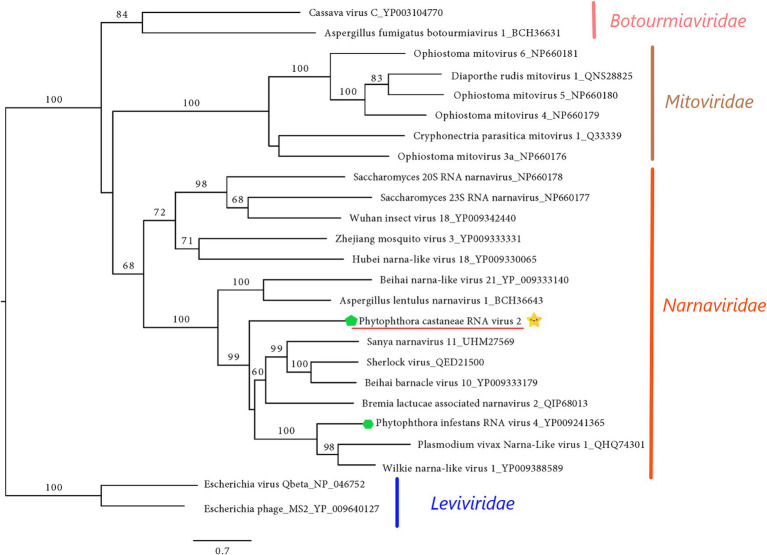
 ) with other (+)ssRNA viruses belonging to families Narnaviridae, Mitoviridae, Leiviviridae, and Botourmiaviridae. The viruses described from oomycetes are indicated by a green hexagone
) with other (+)ssRNA viruses belonging to families Narnaviridae, Mitoviridae, Leiviviridae, and Botourmiaviridae. The viruses described from oomycetes are indicated by a green hexagone  . Nodes are labeled with bootstrap percentages ≥50%. Branch lengths are scaled to the expected underlying number of amino acid substitutions per site. Family classification and the corresponding pBLAST accession numbers are shown next to the virus names. The tree is rooted in the family Leviviridae.
. Nodes are labeled with bootstrap percentages ≥50%. Branch lengths are scaled to the expected underlying number of amino acid substitutions per site. Family classification and the corresponding pBLAST accession numbers are shown next to the virus names. The tree is rooted in the family Leviviridae.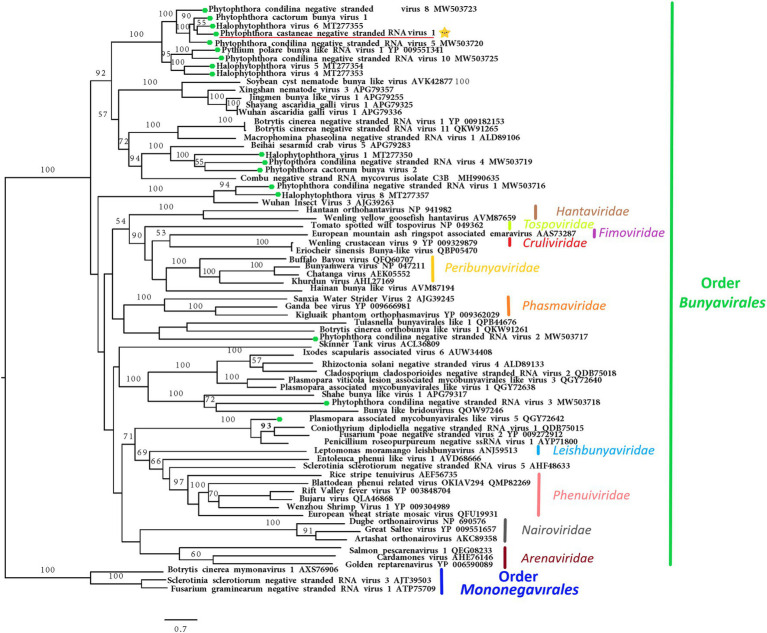
 ) with other complete RdRp belonging to related (−)ssRNA viruses from the orders Bunyavirales and Mononegavirales. The viruses described from oomycetes are indicated by a green hexagone
) with other complete RdRp belonging to related (−)ssRNA viruses from the orders Bunyavirales and Mononegavirales. The viruses described from oomycetes are indicated by a green hexagone  . Nodes are labeled with bootstrap support values. Branch lengths present calculated evolutionary distannce and are scaled to the expected underlying number of amino acid substitutions per site. Nodes are marked with bootstrap percentages ≥50% only. The tree is rooted in Mononegaviales viruses, which are classified within the family Mymonaviridae. Family classification and the corresponding pBLAST accession numbers are shown next to the virus names. The scale bar is indicatig 0.7 aa substitutions per site, per branch.
. Nodes are labeled with bootstrap support values. Branch lengths present calculated evolutionary distannce and are scaled to the expected underlying number of amino acid substitutions per site. Nodes are marked with bootstrap percentages ≥50% only. The tree is rooted in Mononegaviales viruses, which are classified within the family Mymonaviridae. Family classification and the corresponding pBLAST accession numbers are shown next to the virus names. The scale bar is indicatig 0.7 aa substitutions per site, per branch.References
-
- Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Oxford University Press Available at: https://academic.oup.com/nar/article/25/17/3389/1061651 (Accessed January 6, 2021). - PMC - PubMed
-
- Andrews S. (2010). Babraham bioinformatics - FastQC A quality control tool for high throughput sequence data. Soil Available at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (Accessed January 5, 2021).
LinkOut - more resources
Full Text Sources
Research Materials