

Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD): A Review of Clinical and MRI Features, Diagnosis, and Management
- PMID: 35785363
- PMCID: PMC9247462
- DOI: 10.3389/fneur.2022.885218
Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease (MOGAD): A Review of Clinical and MRI Features, Diagnosis, and Management
Abstract
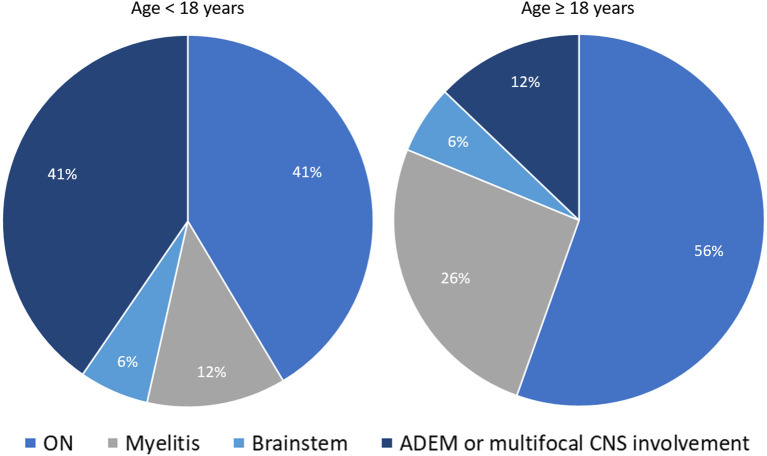
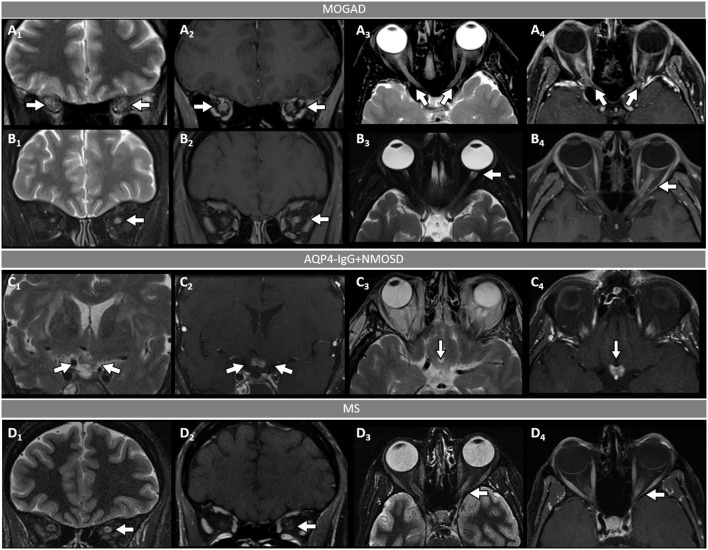
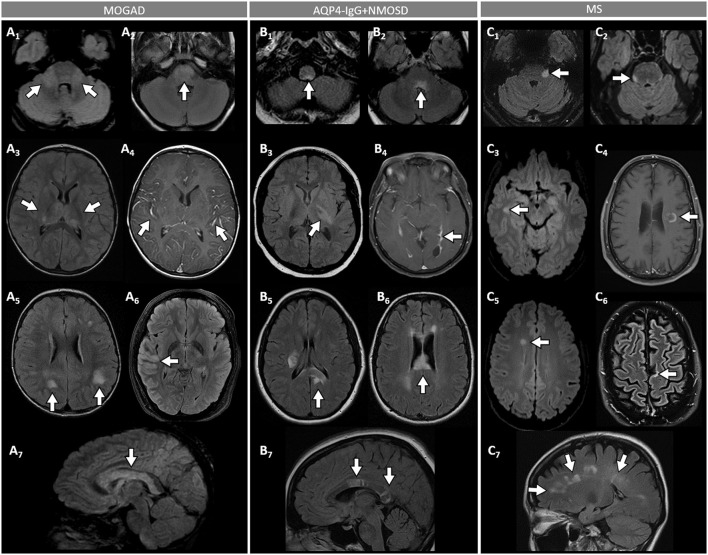
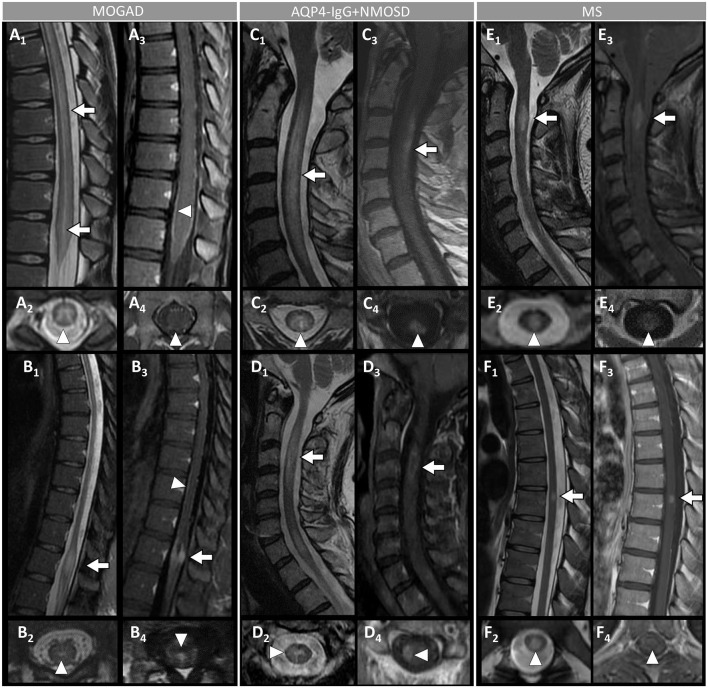
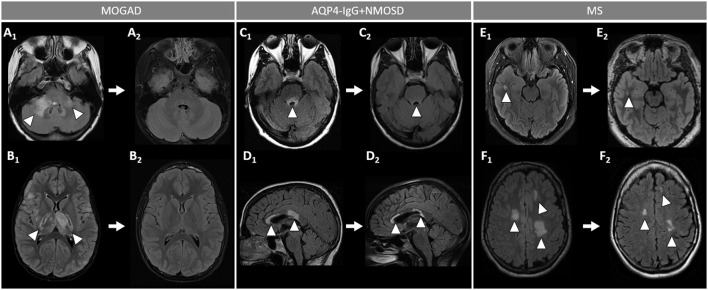
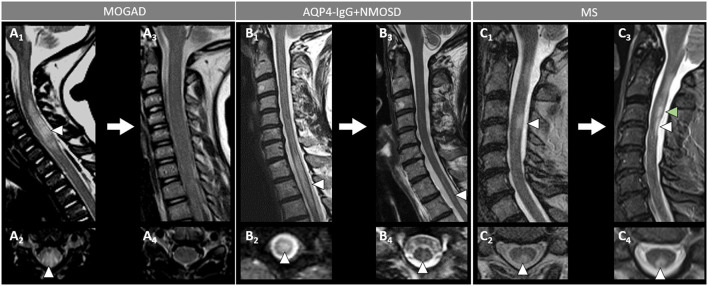
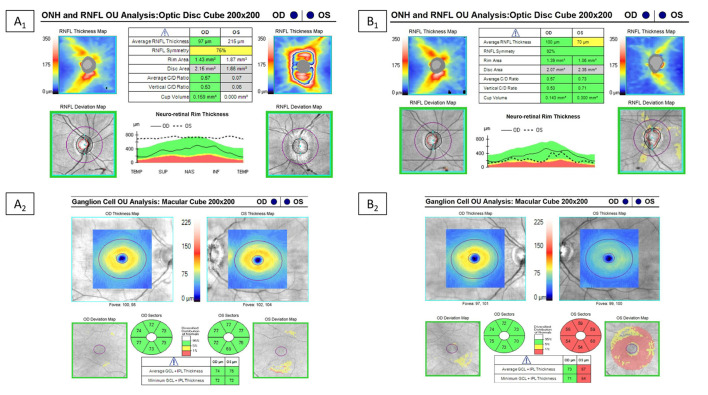
Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease (MOGAD) is the most recently defined inflammatory demyelinating disease of the central nervous system (CNS). Over the last decade, several studies have helped delineate the characteristic clinical-MRI phenotypes of the disease, allowing distinction from aquaporin-4 (AQP4)-IgG-positive neuromyelitis optica spectrum disorder (AQP4-IgG+NMOSD) and multiple sclerosis (MS). The clinical manifestations of MOGAD are heterogeneous, ranging from isolated optic neuritis or myelitis to multifocal CNS demyelination often in the form of acute disseminated encephalomyelitis (ADEM), or cortical encephalitis. A relapsing course is observed in approximately 50% of patients. Characteristic MRI features have been described that increase the diagnostic suspicion (e.g., perineural optic nerve enhancement, spinal cord H-sign, T2-lesion resolution over time) and help discriminate from MS and AQP4+NMOSD, despite some overlap. The detection of MOG-IgG in the serum (and sometimes CSF) confirms the diagnosis in patients with compatible clinical-MRI phenotypes, but false positive results are occasionally encountered, especially with indiscriminate testing of large unselected populations. The type of cell-based assay used to evaluate for MOG-IgG (fixed vs. live) and antibody end-titer (low vs. high) can influence the likelihood of MOGAD diagnosis. International consensus diagnostic criteria for MOGAD are currently being compiled and will assist in clinical diagnosis and be useful for enrolment in clinical trials. Although randomized controlled trials are lacking, MOGAD acute attacks appear to be very responsive to high dose steroids and plasma exchange may be considered in refractory cases. Attack-prevention treatments also lack class-I data and empiric maintenance treatment is generally reserved for relapsing cases or patients with severe residual disability after the presenting attack. A variety of empiric steroid-sparing immunosuppressants can be considered and may be efficacious based on retrospective or prospective observational studies but prospective randomized placebo-controlled trials are needed to better guide treatment. In summary, this article will review our rapidly evolving understanding of MOGAD diagnosis and management.
Keywords: MOG; NMOSD; demyelinating diseases; differential diagnosis; false positive; multiple sclerosis; neuromyelitis optica.
Copyright © 2022 Sechi, Cacciaguerra, Chen, Mariotto, Fadda, Dinoto, Lopez-Chiriboga, Pittock and Flanagan.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources