

Epigenetic modifications in metabolic memory: What are the memories, and can we erase them?
- PMID: 35785987
- PMCID: PMC9359656
- DOI: 10.1152/ajpcell.00201.2022
Epigenetic modifications in metabolic memory: What are the memories, and can we erase them?
Abstract
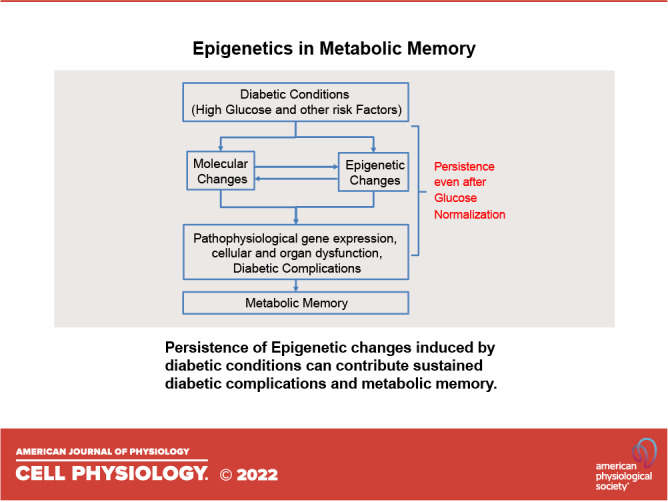
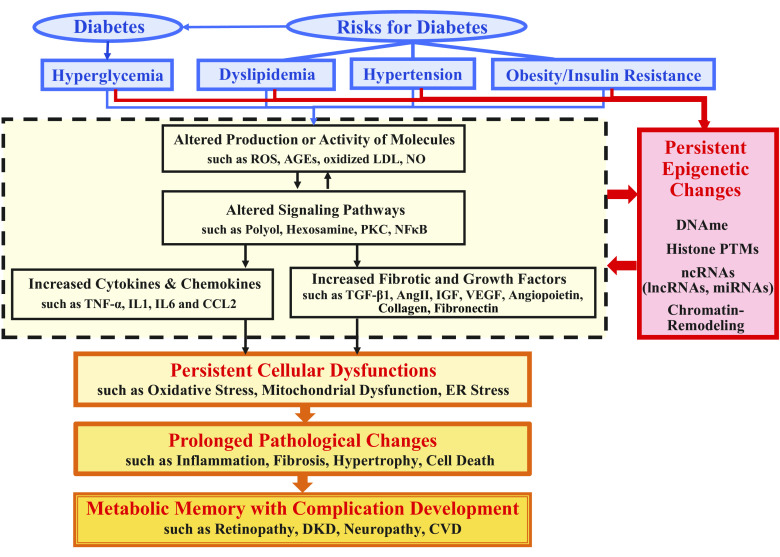
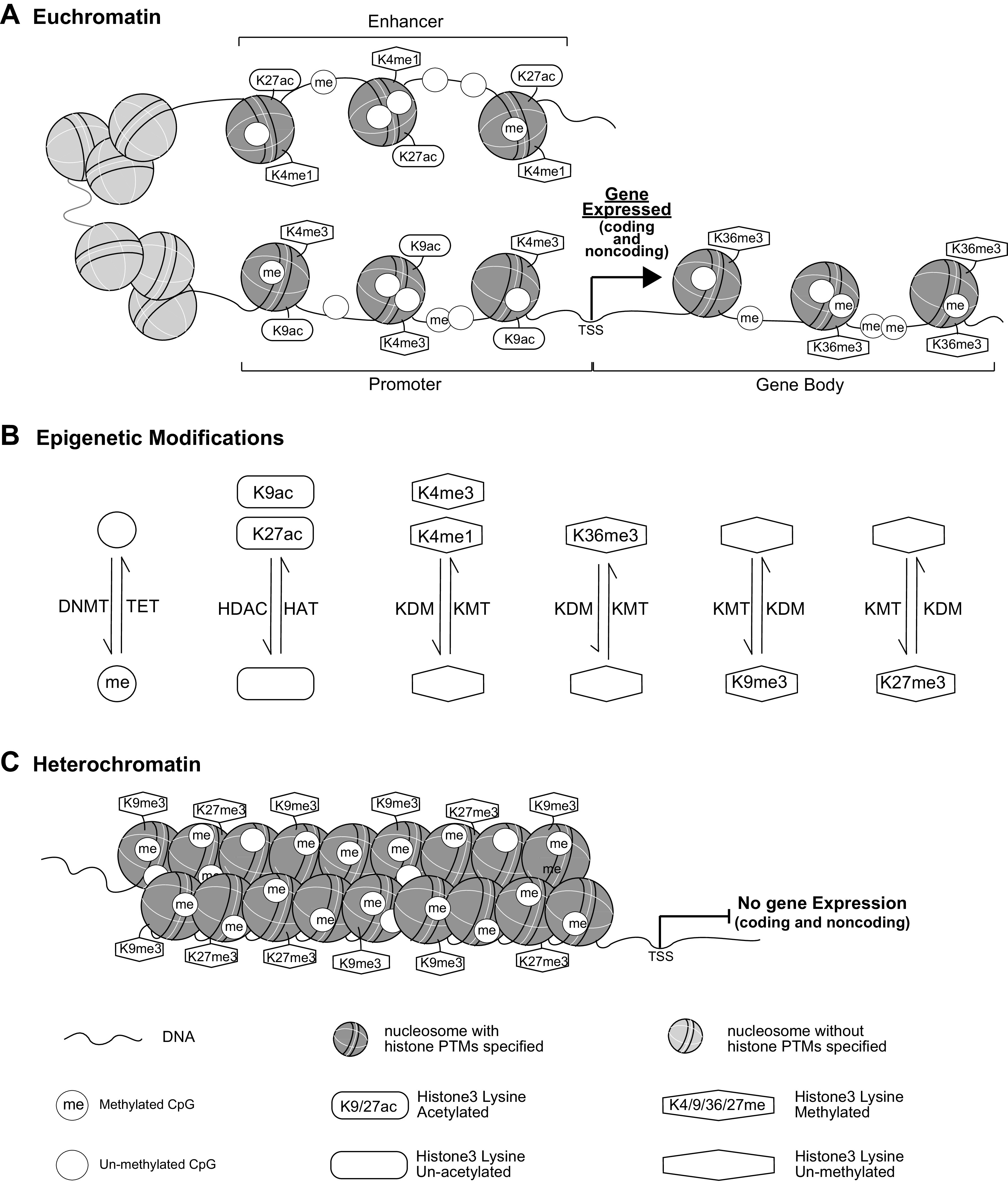
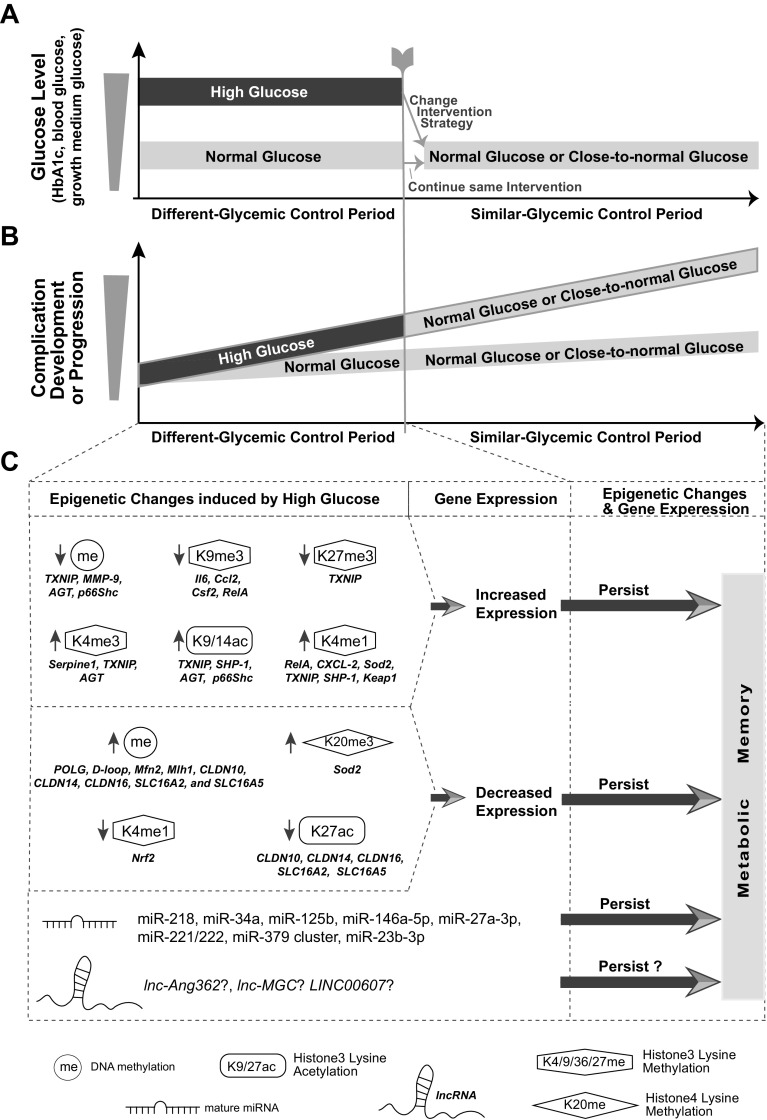
Inherent and acquired abnormalities in gene regulation due to the influence of genetics and epigenetics (traits related to environment rather than genetic factors) underlie many diseases including diabetes. Diabetes could lead to multiple complications including retinopathy, nephropathy, and cardiovascular disease that greatly increase morbidity and mortality. Epigenetic changes have also been linked to diabetes-related complications. Genes associated with many pathophysiological features of these vascular complications (e.g., inflammation, fibrosis, and oxidative stress) can be regulated by epigenetic mechanisms involving histone posttranslational modifications, DNA methylation, changes in chromatin structure/remodeling, and noncoding RNAs. Intriguingly, these epigenetic changes triggered during early periods of hyperglycemic exposure and uncontrolled diabetes are not immediately corrected even after restoration of normoglycemia and metabolic balance. This latency in effect across time and conditions is associated with persistent development of complications in diabetes with prior history of poor glycemic control, termed as metabolic memory or legacy effect. Epigenetic modifications are generally reversible and provide a window of therapeutic opportunity to ameliorate cellular dysfunction and mitigate or "erase" metabolic memory. Notably, trained immunity and related epigenetic changes transmitted from hematopoietic stem cells to innate immune cells have also been implicated in metabolic memory. Hence, identification of epigenetic variations at candidate genes, or epigenetic signatures genome-wide by epigenome-wide association studies can aid in prompt diagnosis to prevent progression of complications and identification of much-needed new therapeutic targets. Herein, we provide a review of epigenetics and epigenomics in metabolic memory of diabetic complications covering the current basic research, clinical data, and translational implications.
Keywords: DNA methylation; diabetic complications; epigenetics; metabolic memory.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
References
-
- Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, Stein C, Basit A, Chan JCN, Mbanya JC, Pavkov ME, Ramachandaran A, Wild SH, James S, Herman WHH, Zhang P, Bommer C, Kuo SH, Boyko EJJ, Magliano DJ. IDF Diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract 183: 109119, 2022. doi: 10.1016/j.diabres.2021.109119. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical