

Multiomic atlas with functional stratification and developmental dynamics of zebrafish cis-regulatory elements
- PMID: 35789323
- PMCID: PMC9279159
- DOI: 10.1038/s41588-022-01089-w
Multiomic atlas with functional stratification and developmental dynamics of zebrafish cis-regulatory elements
Abstract
Zebrafish, a popular organism for studying embryonic development and for modeling human diseases, has so far lacked a systematic functional annotation program akin to those in other animal models. To address this, we formed the international DANIO-CODE consortium and created a central repository to store and process zebrafish developmental functional genomic data. Our data coordination center ( https://danio-code.zfin.org ) combines a total of 1,802 sets of unpublished and re-analyzed published genomic data, which we used to improve existing annotations and show its utility in experimental design. We identified over 140,000 cis-regulatory elements throughout development, including classes with distinct features dependent on their activity in time and space. We delineated the distinct distance topology and chromatin features between regulatory elements active during zygotic genome activation and those active during organogenesis. Finally, we matched regulatory elements and epigenomic landscapes between zebrafish and mouse and predicted functional relationships between them beyond sequence similarity, thus extending the utility of zebrafish developmental genomics to mammals.
© 2022. Crown.
Conflict of interest statement
The authors declare no competing interests.
Figures
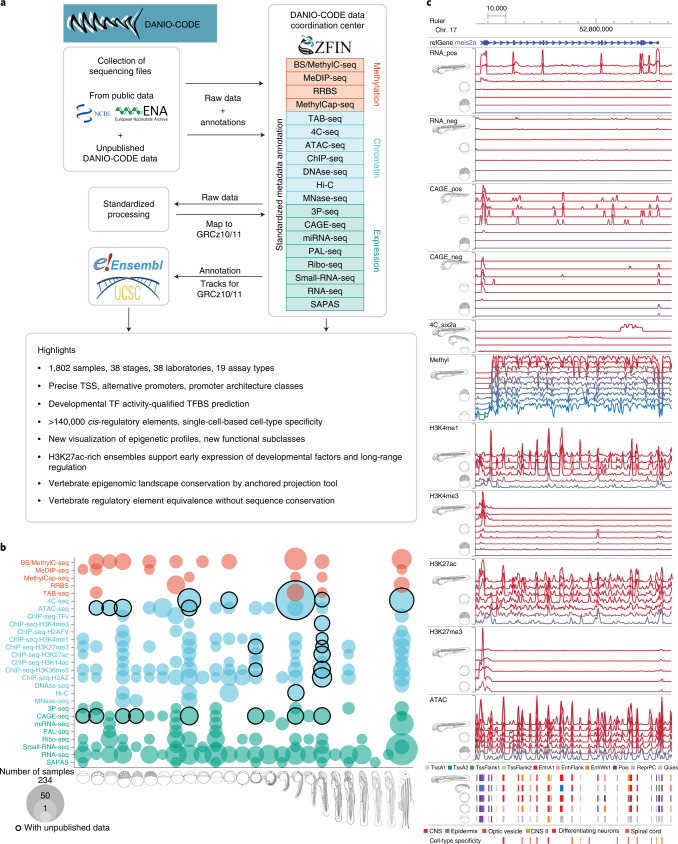





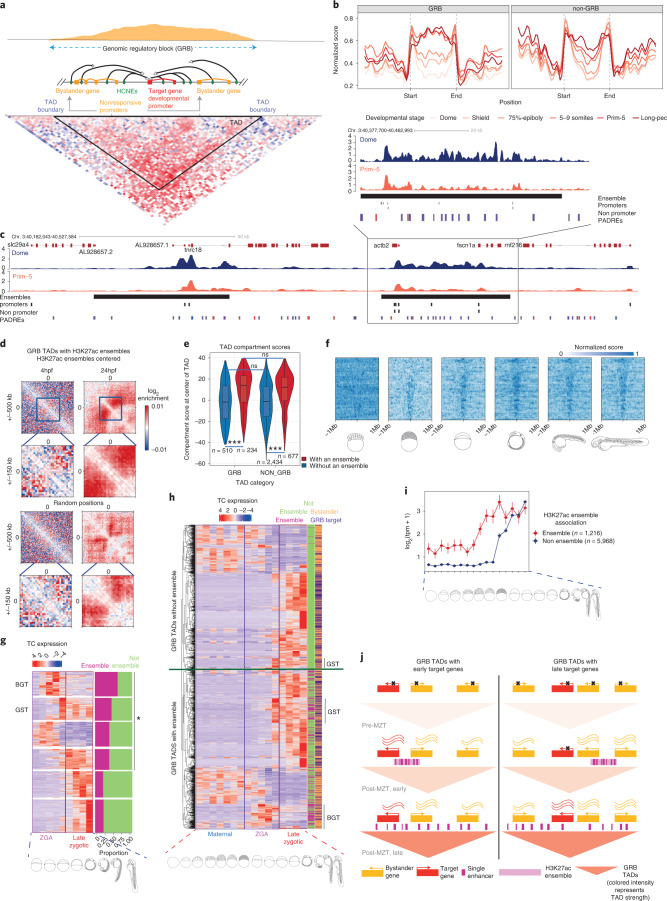
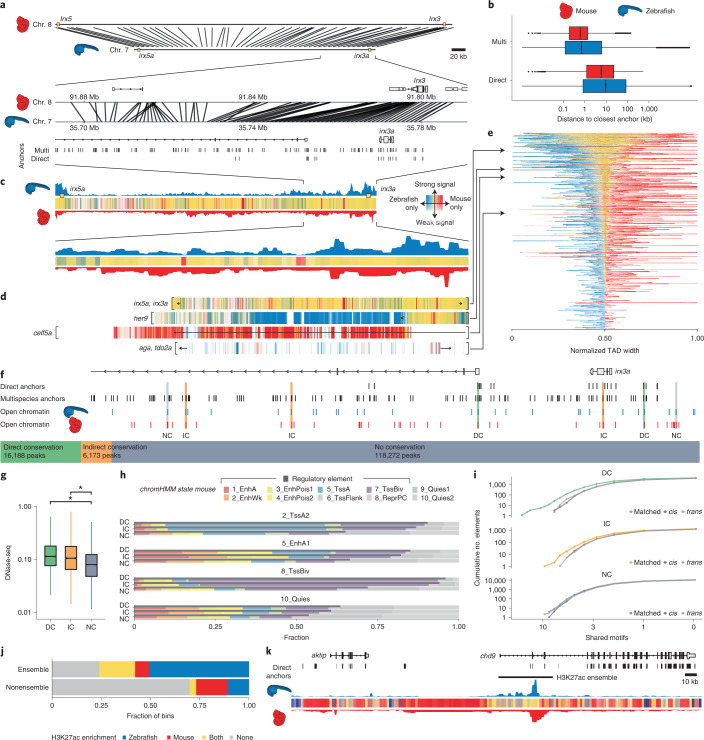










Comment in
-
Decoding the zebrafish genome.Nat Genet. 2022 Jul;54(7):917-919. doi: 10.1038/s41588-022-01080-5. Nat Genet. 2022. PMID: 35789322 No abstract available.
References
-
- Murphy PJ, Wu SF, James CR, Wike CL, Cairns BR. Placeholder nucleosomes underlie germline-to-embryo DNA methylation reprogramming. Cell. 2018;172:993–1006.e13. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U24 HG002659/HG/NHGRI NIH HHS/United States
- BB/R015457/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- MC_EX_MR/S300007/1/MRC_/Medical Research Council/United Kingdom
- 106955/WT_/Wellcome Trust/United Kingdom
- P61715/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
