

BayesR3 enables fast MCMC blocked processing for largescale multi-trait genomic prediction and QTN mapping analysis
- PMID: 35790806
- PMCID: PMC9256732
- DOI: 10.1038/s42003-022-03624-1
BayesR3 enables fast MCMC blocked processing for largescale multi-trait genomic prediction and QTN mapping analysis
Abstract
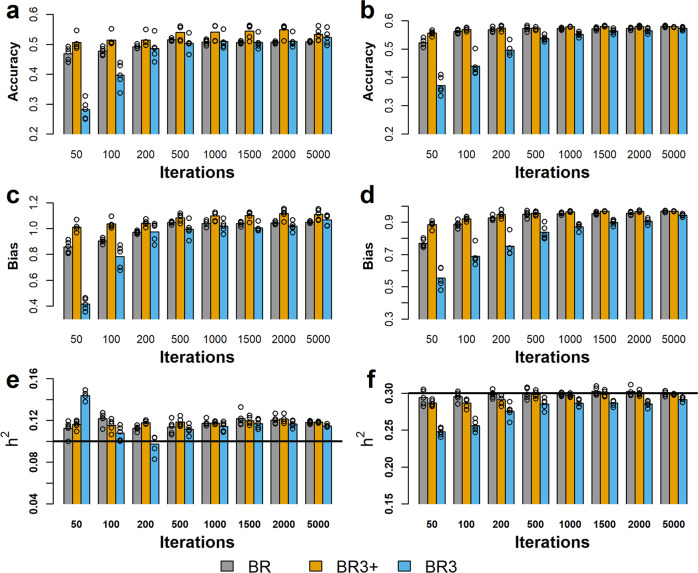
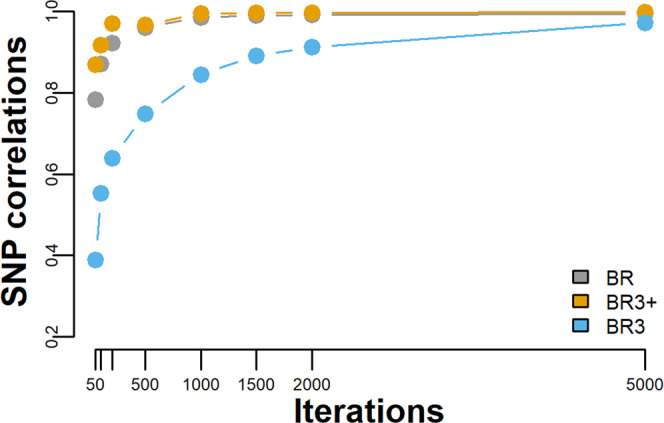
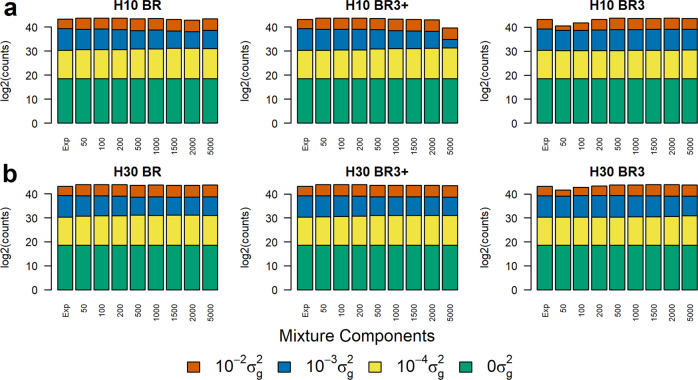
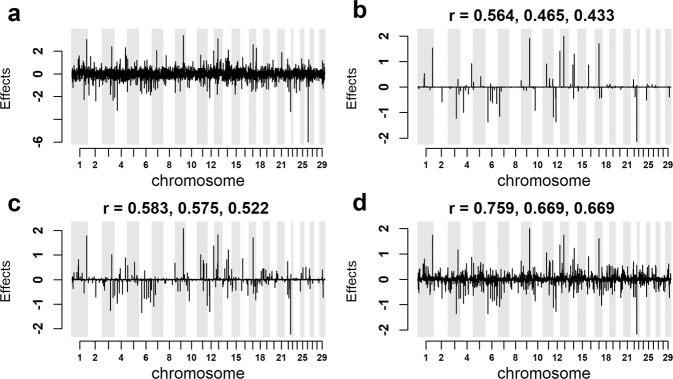
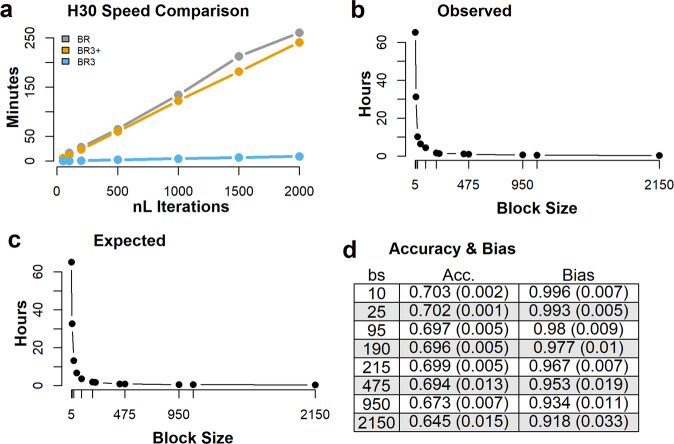
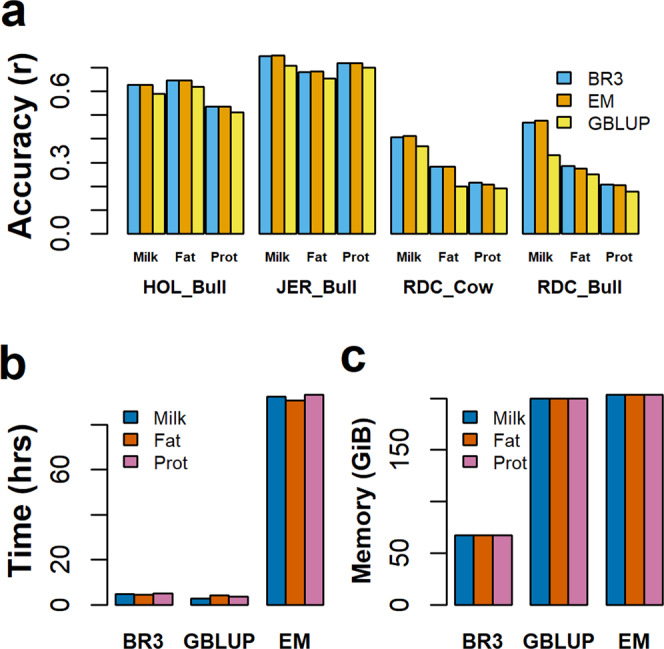
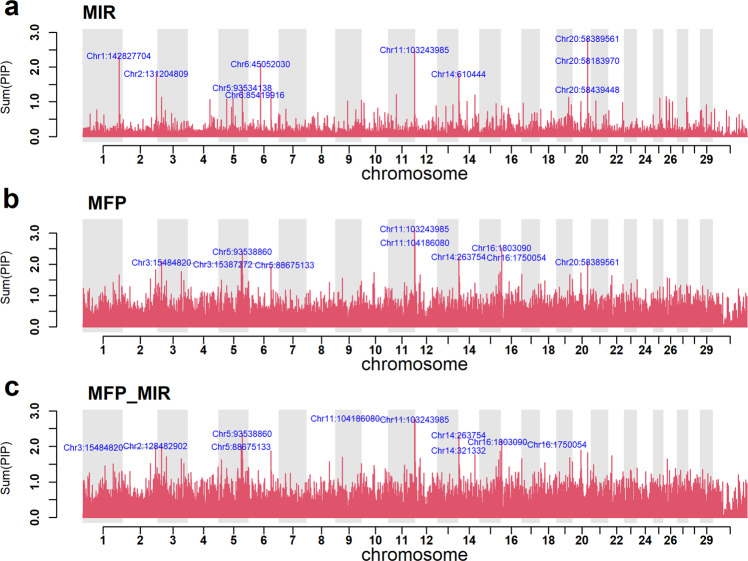
Bayesian methods, such as BayesR, for predicting the genetic value or risk of individuals from their genotypes, such as Single Nucleotide Polymorphisms (SNP), are often implemented using a Markov Chain Monte Carlo (MCMC) process. However, the generation of Markov chains is computationally slow. We introduce a form of blocked Gibbs sampling for estimating SNP effects from Markov chains that greatly reduces computational time by sampling each SNP effect iteratively n-times from conditional block posteriors. Subsequent iteration over all blocks m-times produces chains of length m × n. We use this strategy to solve large-scale genomic prediction and fine-mapping problems using the Bayesian MCMC mixed-effects genetic model, BayesR3. We validate the method using simulated data, followed by analysis of empirical dairy cattle data using high dimension milk mid infra-red spectra data as an example of "omics" data and show its use to increase the precision of mapping variants affecting milk, fat, and protein yields relative to a univariate analysis of milk, fat, and protein.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Timpson NJ, Greenwood CMT, Soranzo N, Lawson DJ, Richards JB. Genetic architecture: the shape of the genetic contribution to human traits and disease. Nat. Rev. Genetics. 2018;19:110–124. - PubMed
-
- Visscher PM, Yang J, Goddard ME. A commentary on ‘common SNPs explain a large proportion of the heritability for human height’ by Yang et al. (2010) Twin Res Hum. Genetics. 2010;13:517–524. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
