

Signal amplification in the KEAP1-NRF2-ARE antioxidant response pathway
- PMID: 35792437
- PMCID: PMC9287733
- DOI: 10.1016/j.redox.2022.102389
Signal amplification in the KEAP1-NRF2-ARE antioxidant response pathway
Abstract
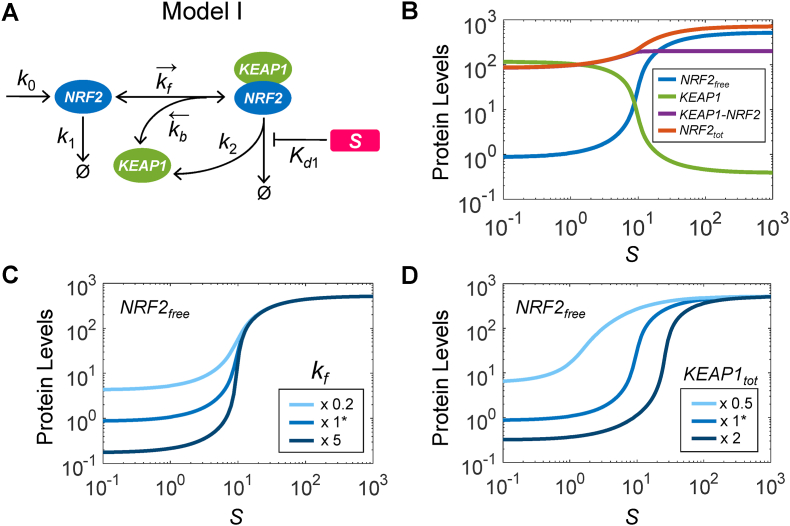
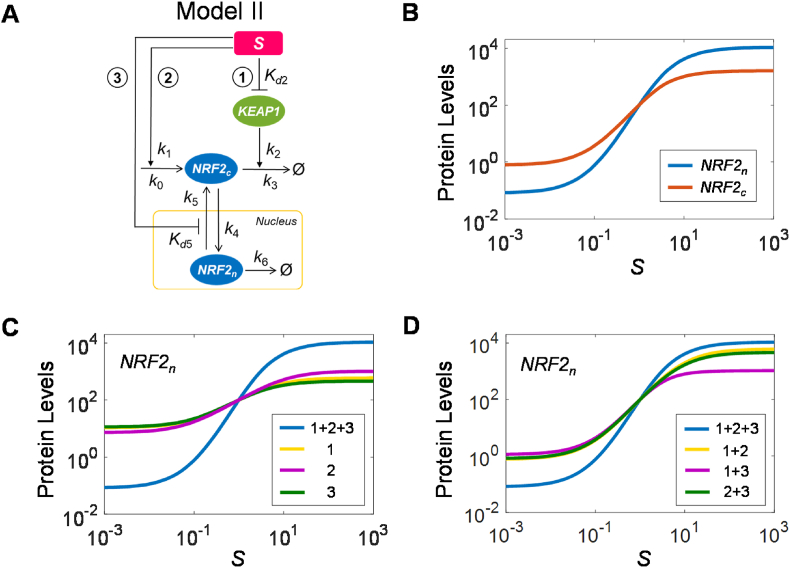
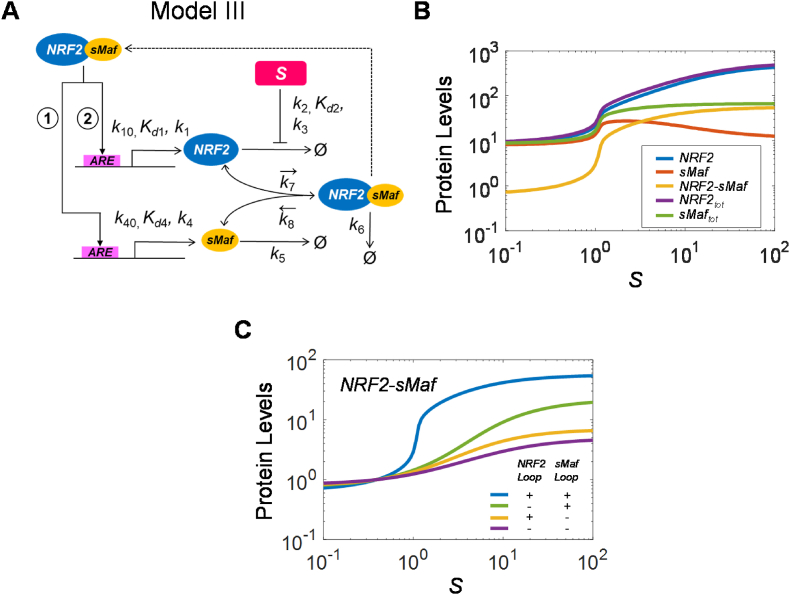
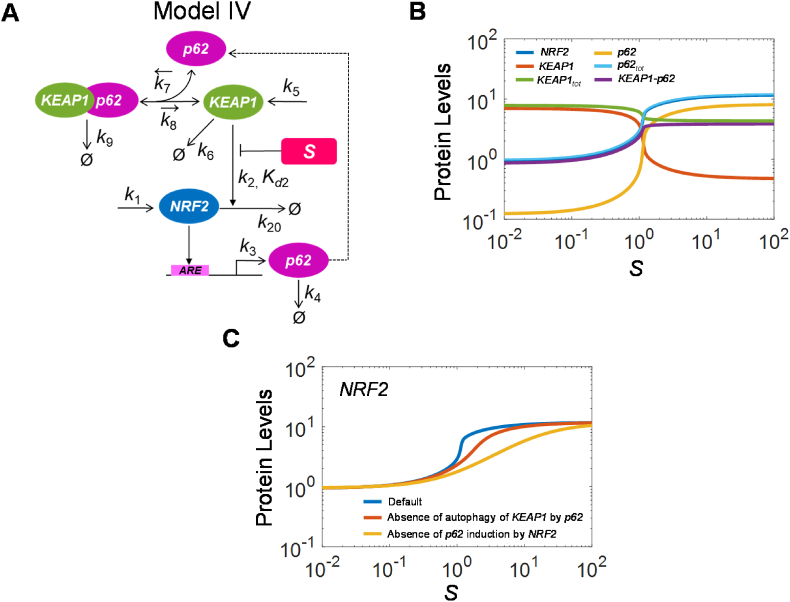
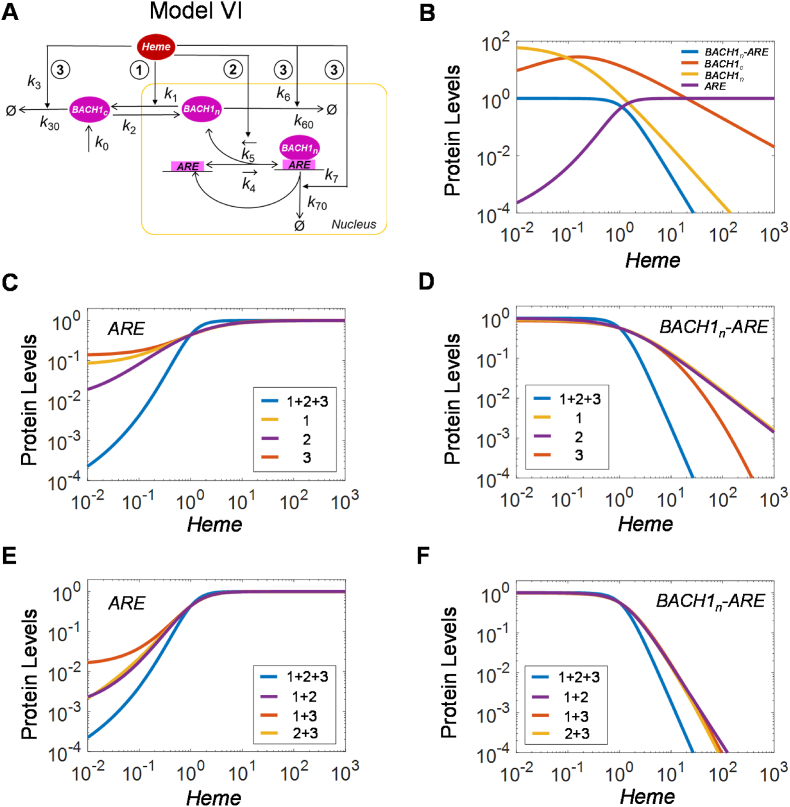
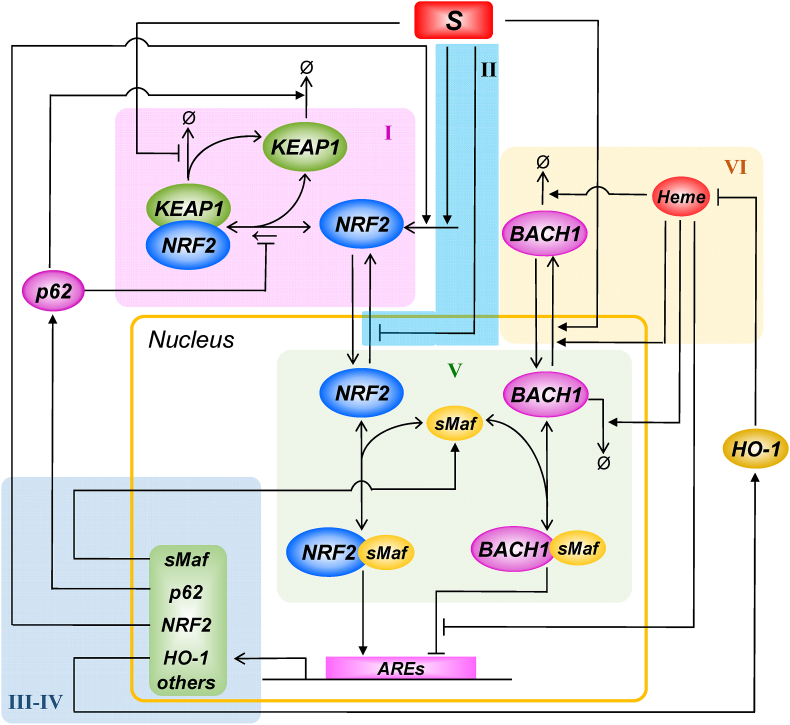
The KEAP1-NRF2-ARE signaling pathway plays a central role in mediating the adaptive cellular stress response to oxidative and electrophilic chemicals. This canonical pathway has been extensively studied and reviewed in the past two decades, but rarely was it looked at from a quantitative signaling perspective. Signal amplification, i.e., ultrasensitivity, is crucially important for robust induction of antioxidant genes to appropriate levels that can adequately counteract the stresses. In this review article, we examined a number of well-known molecular events in the KEAP1-NRF2-ARE pathway from a quantitative perspective with a focus on how signal amplification can be achieved. We illustrated, by using a series of mathematical models, that redox-regulated protein sequestration, stabilization, translation, nuclear trafficking, DNA promoter binding, and transcriptional induction - which are embedded in the molecular network comprising KEAP1, NRF2, sMaf, p62, and BACH1 - may generate highly ultrasensitive NRF2 activation and antioxidant gene induction. The emergence and degree of ultrasensitivity depend on the strengths of protein-protein and protein-DNA interaction and protein abundances. A unique, quantitative understanding of signal amplification in the KEAP1-NRF2-ARE pathway will help to identify sensitive targets for the prevention and therapeutics of oxidative stress-related diseases and develop quantitative adverse outcome pathway models to facilitate the health risk assessment of oxidative chemicals.
Keywords: ARE; KEAP1; NRF2; Oxidative stress; Signal amplification; Ultrasensitivity.
Copyright © 2022 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Nguyen T., Sherratt P.J., Pickett C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003;43:233–260. - PubMed
-
- Torrente L., DeNicola G.M. Targeting NRF2 and its downstream processes: opportunities and challenges. Annu. Rev. Pharmacol. Toxicol. 2022;62:279–300. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
