

Biological Misinterpretation of Transcriptional Signatures in Tumor Samples Can Unknowingly Undermine Mechanistic Understanding and Faithful Alignment with Preclinical Data
- PMID: 35792866
- PMCID: PMC9475248
- DOI: 10.1158/1078-0432.CCR-22-1102
Biological Misinterpretation of Transcriptional Signatures in Tumor Samples Can Unknowingly Undermine Mechanistic Understanding and Faithful Alignment with Preclinical Data
Abstract
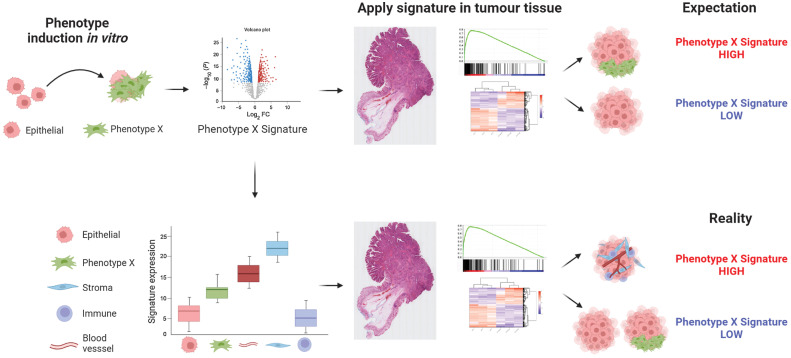
Purpose: Precise mechanism-based gene expression signatures (GES) have been developed in appropriate in vitro and in vivo model systems, to identify important cancer-related signaling processes. However, some GESs originally developed to represent specific disease processes, primarily with an epithelial cell focus, are being applied to heterogeneous tumor samples where the expression of the genes in the signature may no longer be epithelial-specific. Therefore, unknowingly, even small changes in tumor stroma percentage can directly influence GESs, undermining the intended mechanistic signaling.
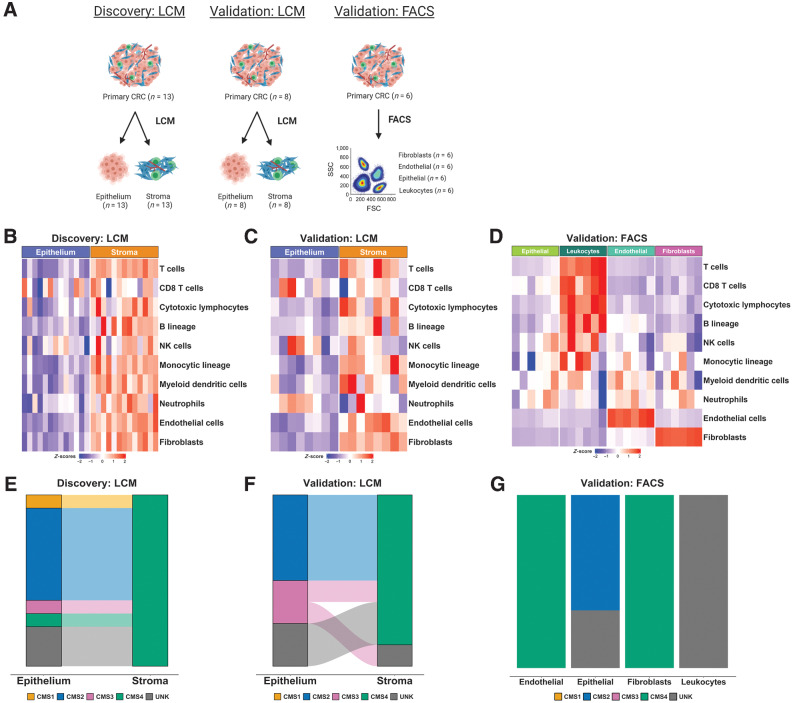
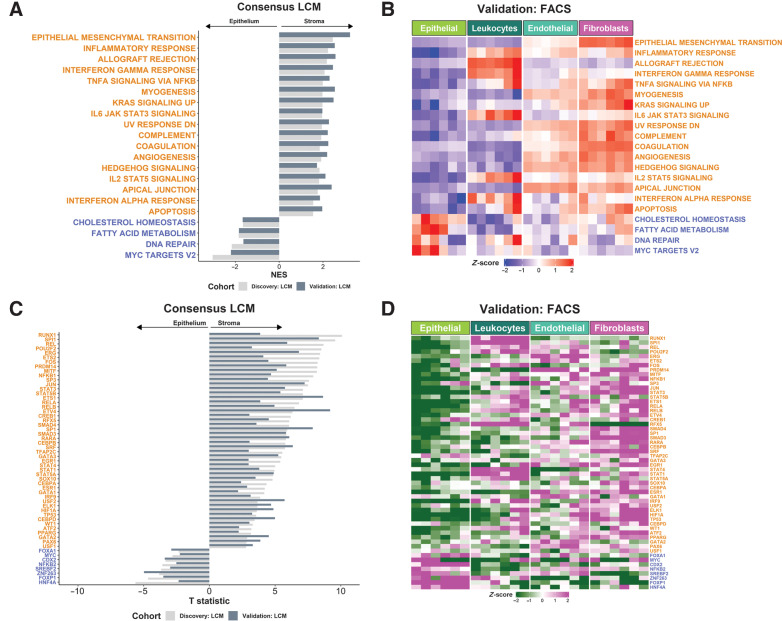
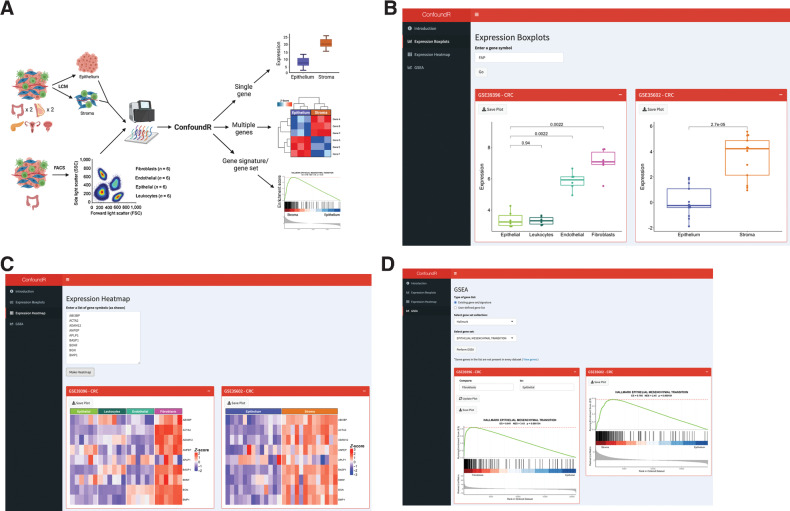
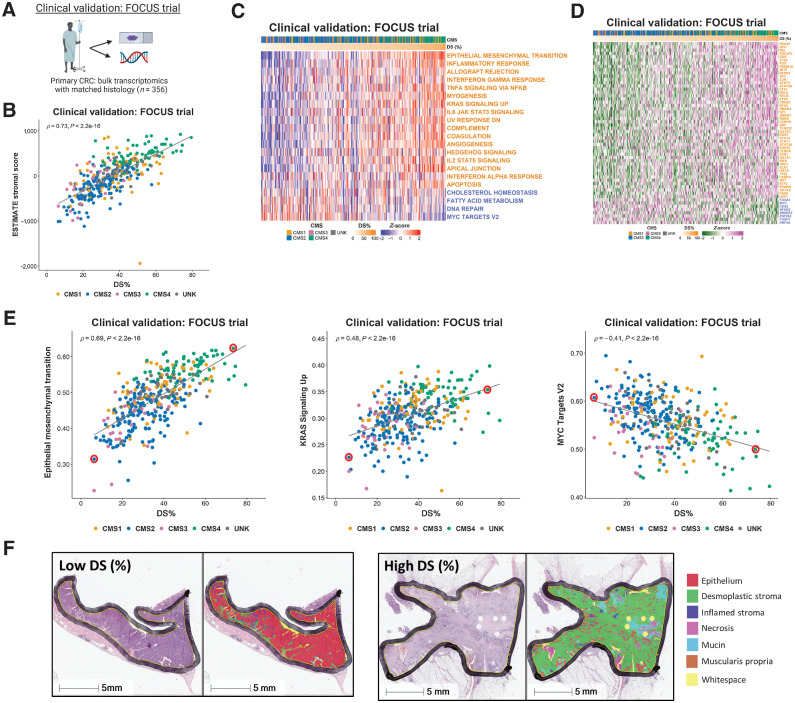
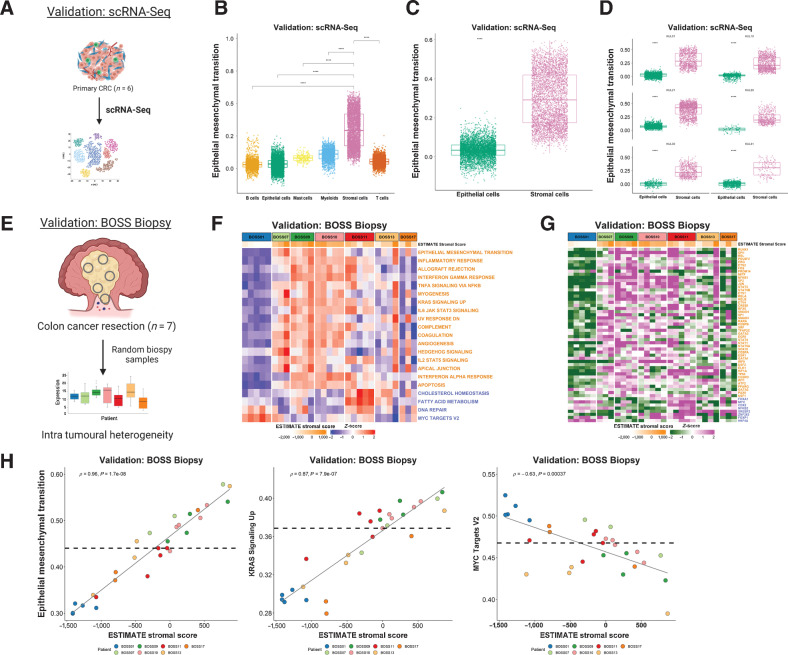
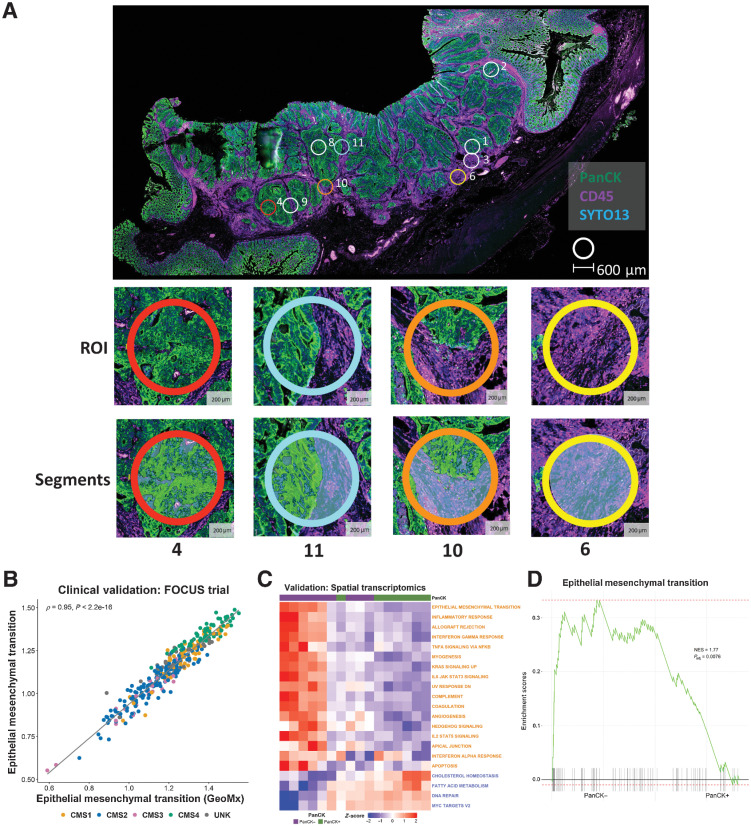
Experimental design: Using colorectal cancer as an exemplar, we deployed numerous orthogonal profiling methodologies, including laser capture microdissection, flow cytometry, bulk and multiregional biopsy clinical samples, single-cell RNA sequencing and finally spatial transcriptomics, to perform a comprehensive assessment of the potential for the most widely used GESs to be influenced, or confounded, by stromal content in tumor tissue. To complement this work, we generated a freely-available resource, ConfoundR; https://confoundr.qub.ac.uk/, that enables users to test the extent of stromal influence on an unlimited number of the genes/signatures simultaneously across colorectal, breast, pancreatic, ovarian and prostate cancer datasets.
Results: Findings presented here demonstrate the clear potential for misinterpretation of the meaning of GESs, due to widespread stromal influences, which in-turn can undermine faithful alignment between clinical samples and preclinical data/models, particularly cell lines and organoids, or tumor models not fully recapitulating the stromal and immune microenvironment.
Conclusions: Efforts to faithfully align preclinical models of disease using phenotypically-designed GESs must ensure that the signatures themselves remain representative of the same biology when applied to clinical samples.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet 2015;47:312–9. - PubMed
Publication types
MeSH terms
Grants and funding
- MR/V029711/1/MRC_/Medical Research Council/United Kingdom
- A28223/CRUK_/Cancer Research UK/United Kingdom
- A29834/CRUK_/Cancer Research UK/United Kingdom
- 29834/CRUK_/Cancer Research UK/United Kingdom
- A25142/CRUK_/Cancer Research UK/United Kingdom
- C55370/A25813/CRUK_/Cancer Research UK/United Kingdom
- MR/M016587/1/MRC_/Medical Research Council/United Kingdom
- MC_PC_21042/MRC_/Medical Research Council/United Kingdom
- A26825/CRUK_/Cancer Research UK/United Kingdom
- 168322/SNSF_/Swiss National Science Foundation/Switzerland
- 206314/Z/17/Z/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
