

A time-resolved, multi-symbol molecular recorder via sequential genome editing
- PMID: 35794474
- PMCID: PMC9352581
- DOI: 10.1038/s41586-022-04922-8
A time-resolved, multi-symbol molecular recorder via sequential genome editing
Abstract
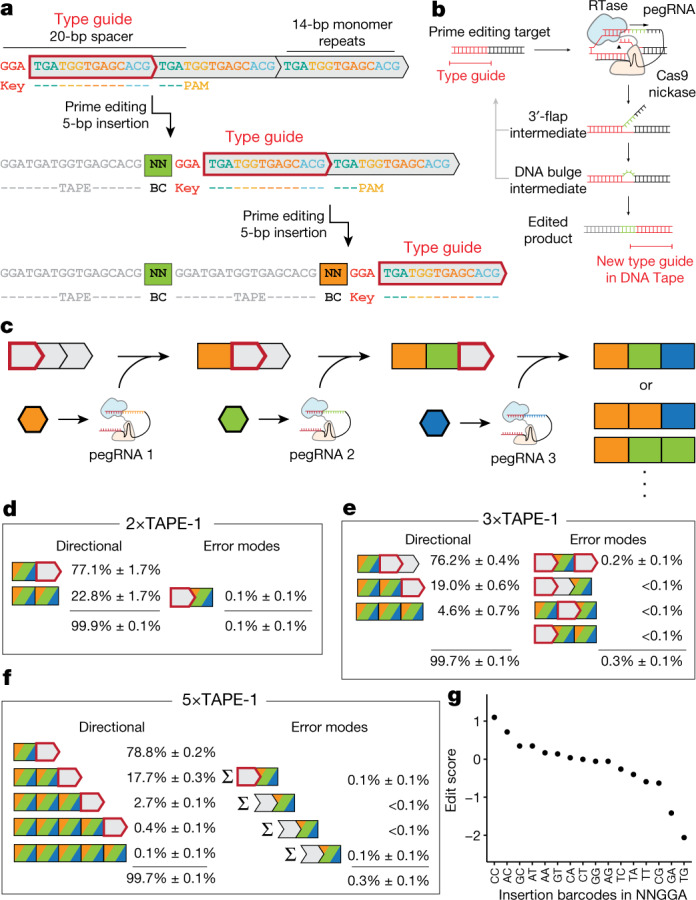
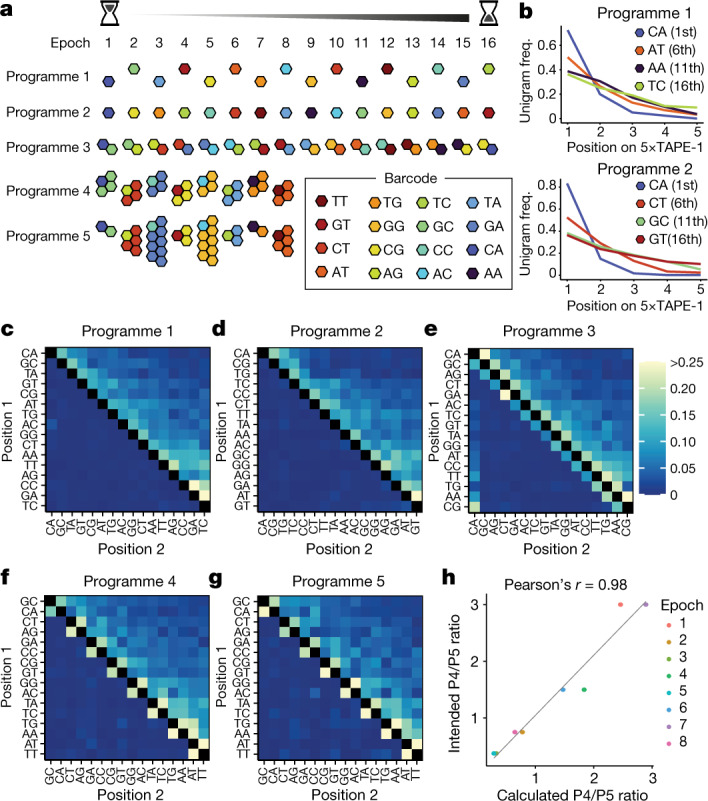
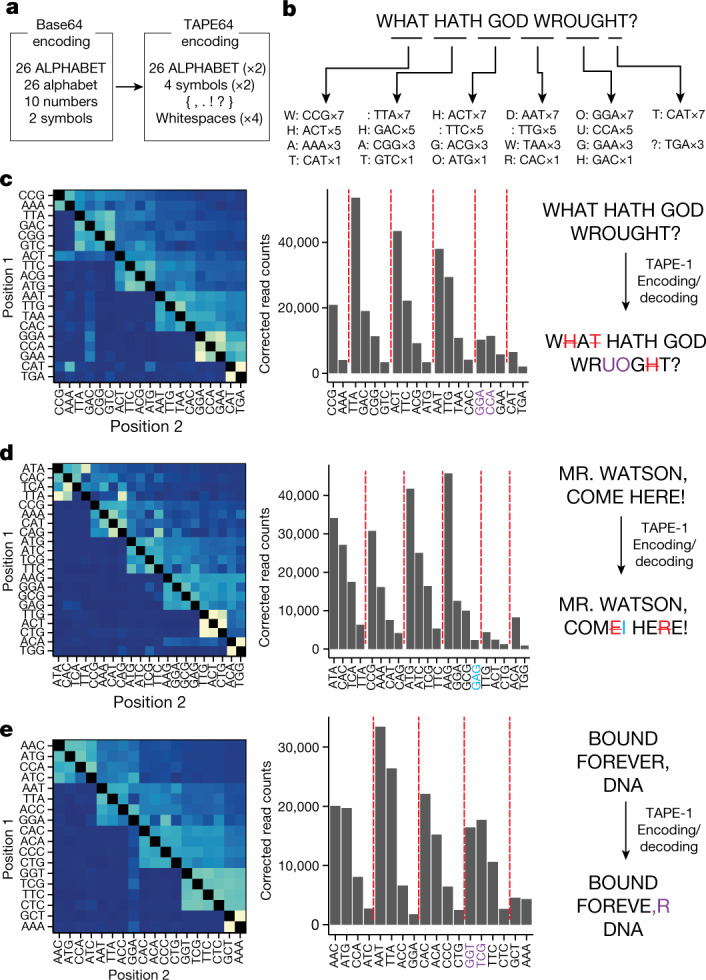
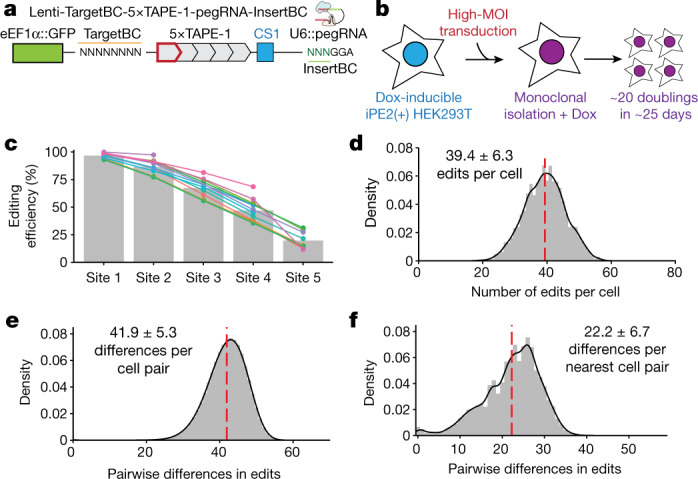
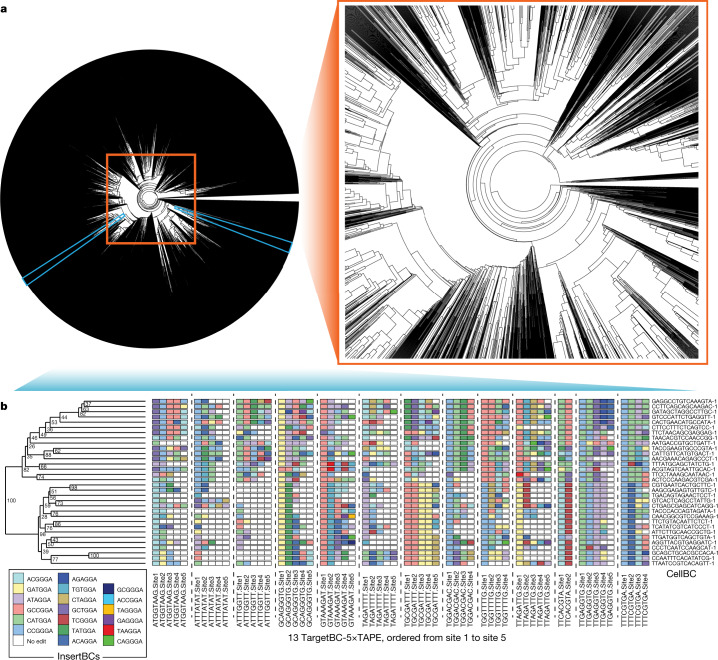
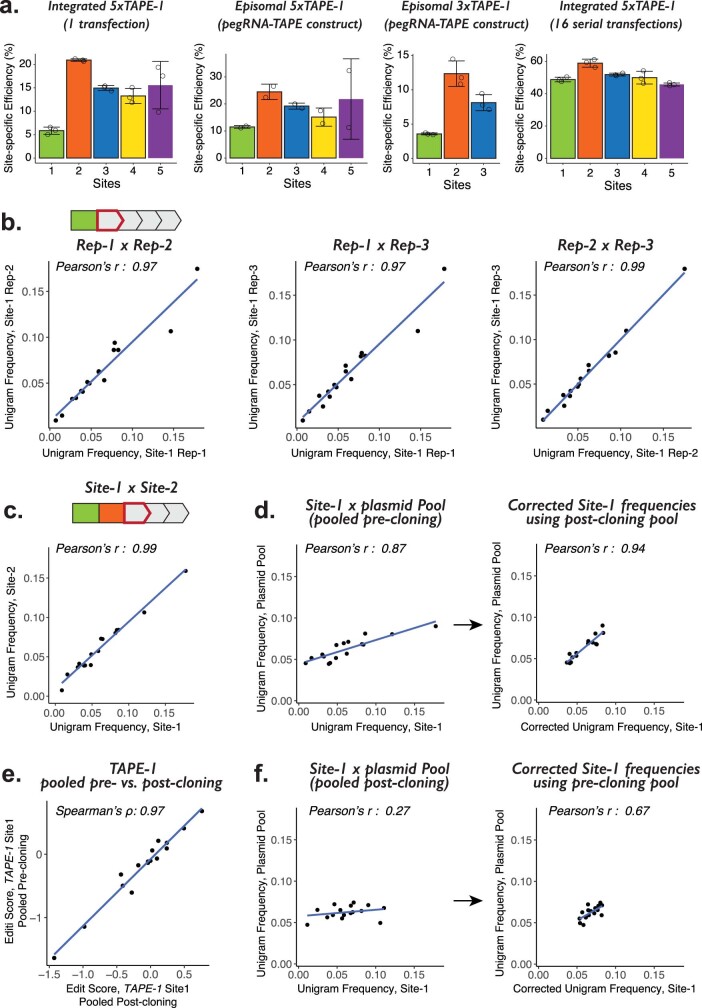
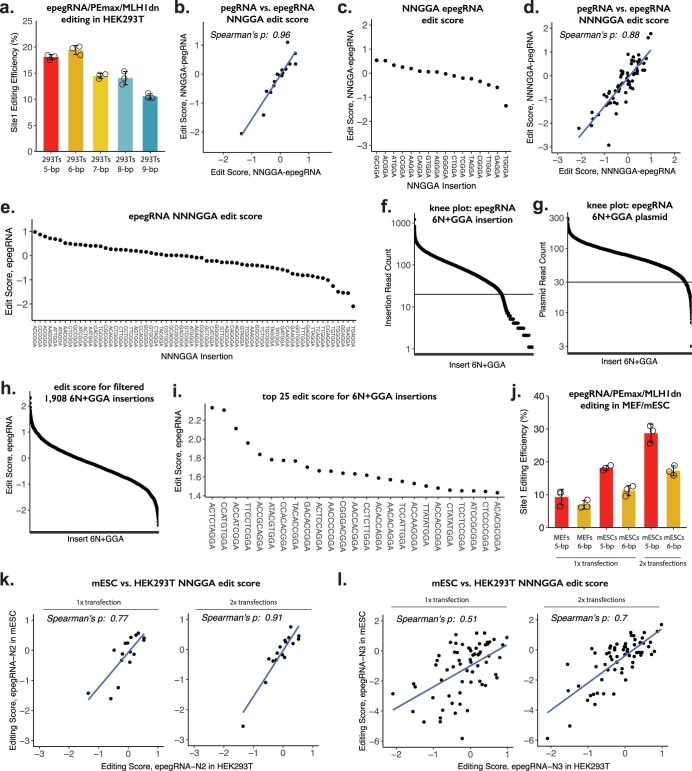
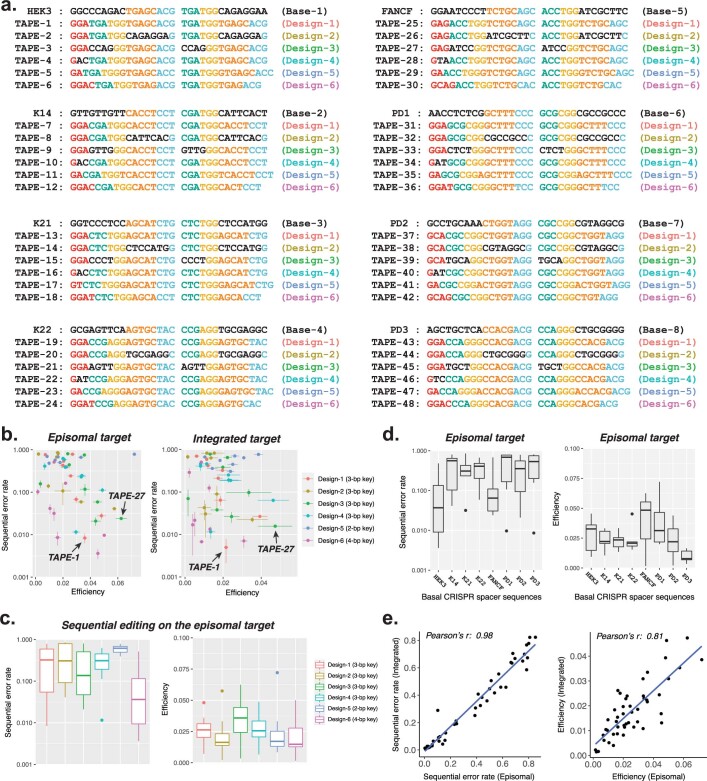
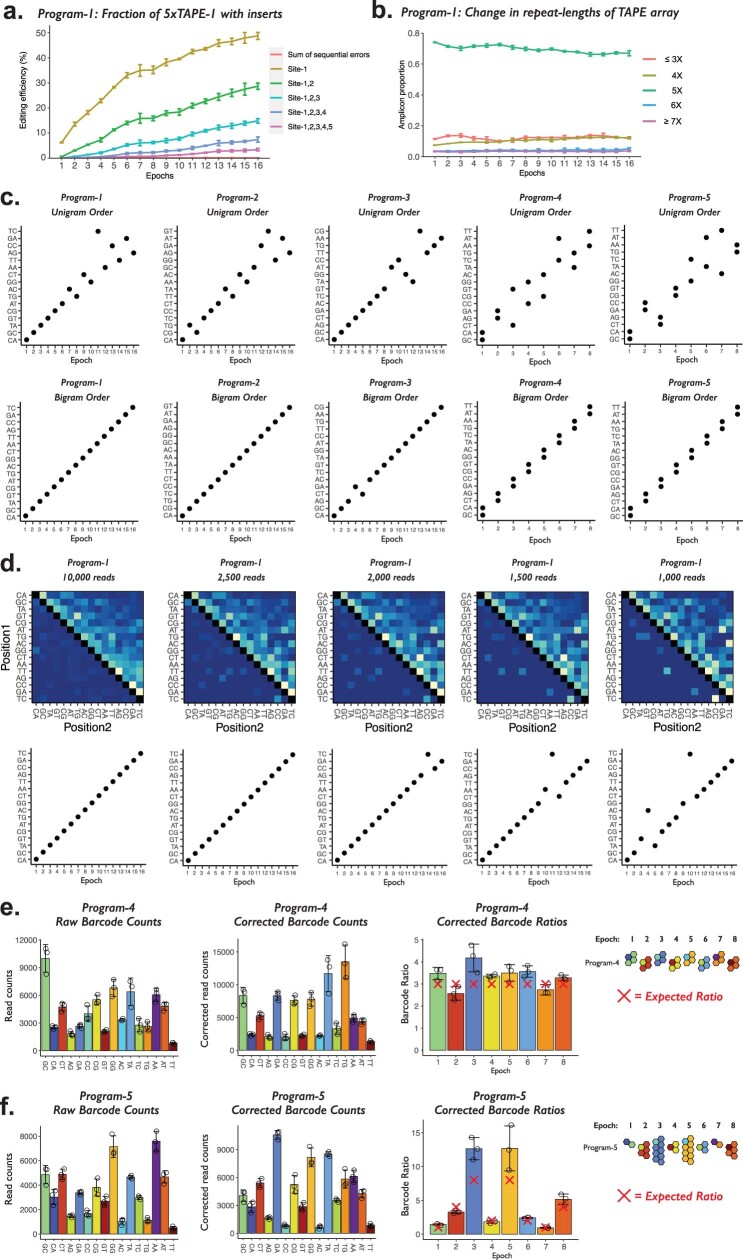
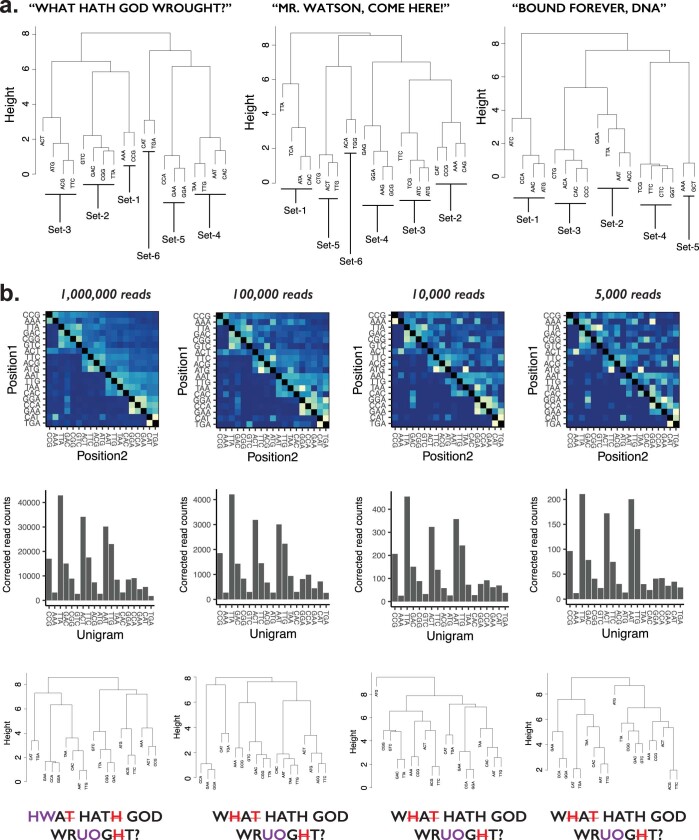
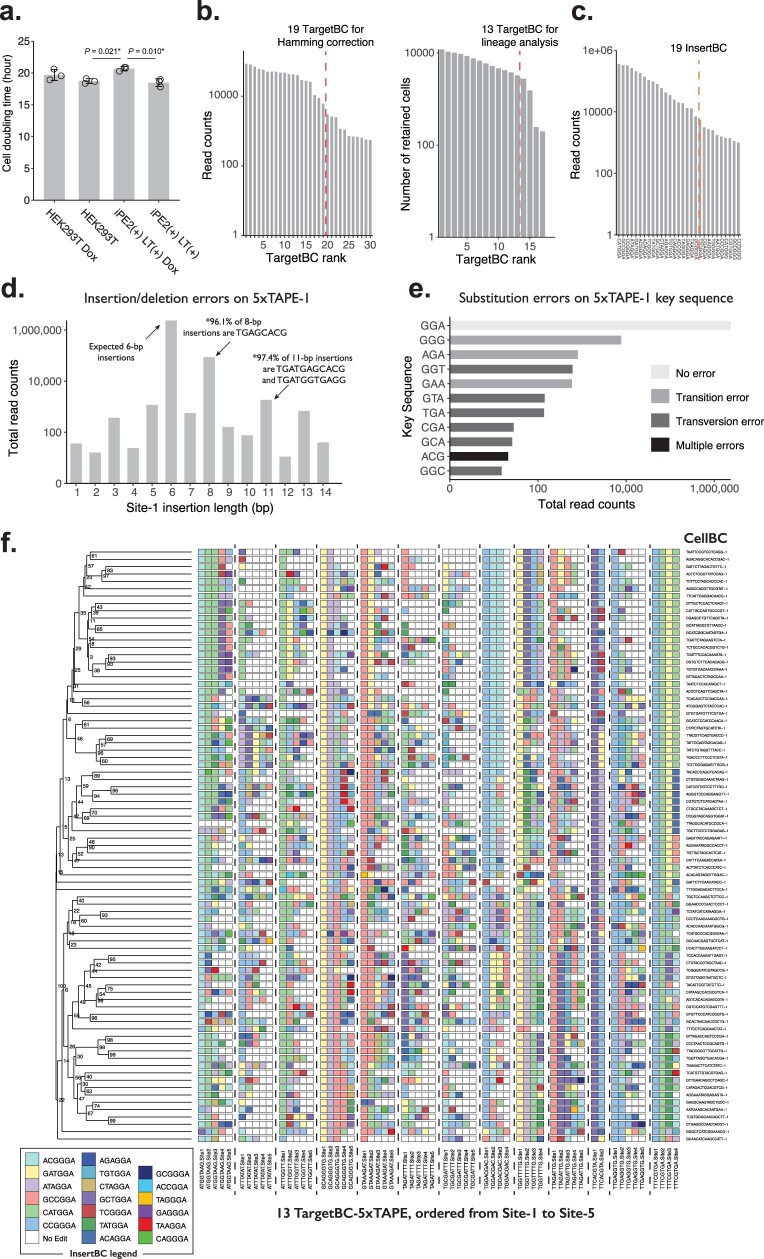
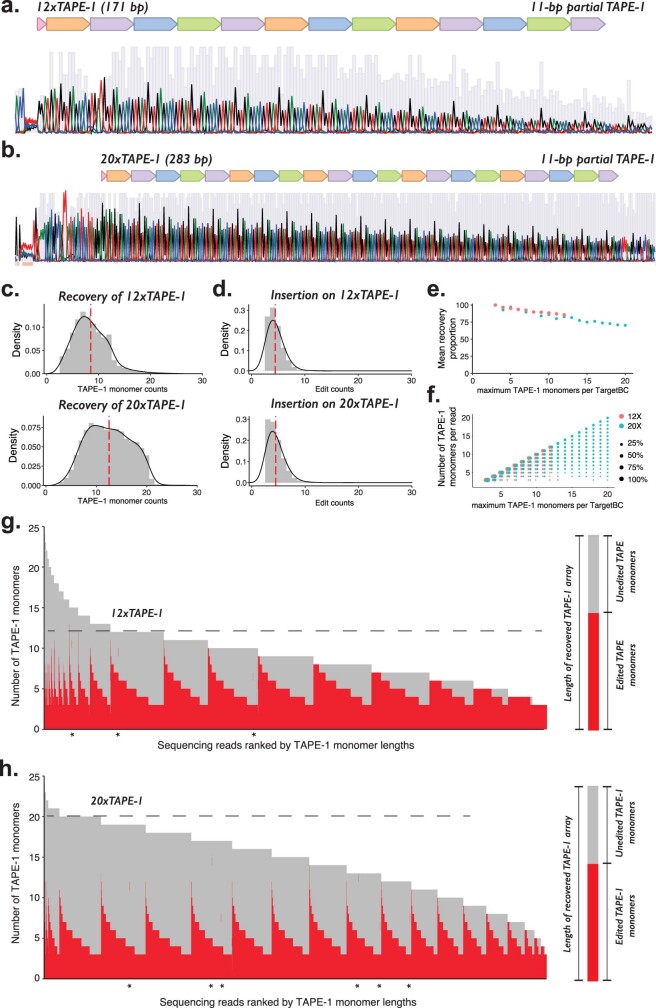
DNA is naturally well suited to serve as a digital medium for in vivo molecular recording. However, contemporary DNA-based memory devices are constrained in terms of the number of distinct 'symbols' that can be concurrently recorded and/or by a failure to capture the order in which events occur1. Here we describe DNA Typewriter, a general system for in vivo molecular recording that overcomes these and other limitations. For DNA Typewriter, the blank recording medium ('DNA Tape') consists of a tandem array of partial CRISPR-Cas9 target sites, with all but the first site truncated at their 5' ends and therefore inactive. Short insertional edits serve as symbols that record the identity of the prime editing guide RNA2 mediating the edit while also shifting the position of the 'type guide' by one unit along the DNA Tape, that is, sequential genome editing. In this proof of concept of DNA Typewriter, we demonstrate recording and decoding of thousands of symbols, complex event histories and short text messages; evaluate the performance of dozens of orthogonal tapes; and construct 'long tape' potentially capable of recording as many as 20 serial events. Finally, we leverage DNA Typewriter in conjunction with single-cell RNA-seq to reconstruct a monophyletic lineage of 3,257 cells and find that the Poisson-like accumulation of sequential edits to multicopy DNA tape can be maintained across at least 20 generations and 25 days of in vitro clonal expansion.
© 2022. The Author(s).
Conflict of interest statement
The University of Washington has filed a patent application partially based on this work in which J.C., W.C. and J.S. are listed as inventors. The remaining authors declare no competing interests.
Figures
Comment in
-
DNA Typewriter.Nat Rev Genet. 2022 Sep;23(9):521. doi: 10.1038/s41576-022-00523-3. Nat Rev Genet. 2022. PMID: 35869289 No abstract available.
References
-
- Church, G. & Shendure, J. Nucleic acid memory device. US patent US20100099080A1 (2003).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
