

A transcriptomic axis predicts state modulation of cortical interneurons
- PMID: 35794483
- PMCID: PMC9279161
- DOI: 10.1038/s41586-022-04915-7
A transcriptomic axis predicts state modulation of cortical interneurons
Erratum in
-
Publisher Correction: A transcriptomic axis predicts state modulation of cortical interneurons.Nature. 2022 Sep;609(7927):E10. doi: 10.1038/s41586-022-05209-8. Nature. 2022. PMID: 36008728 Free PMC article. No abstract available.
Abstract
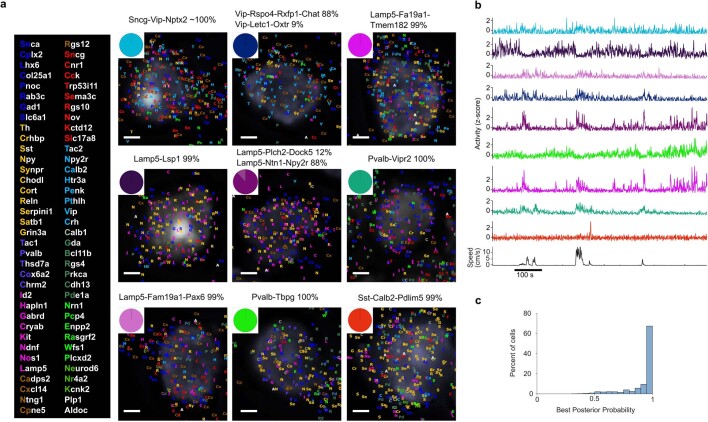
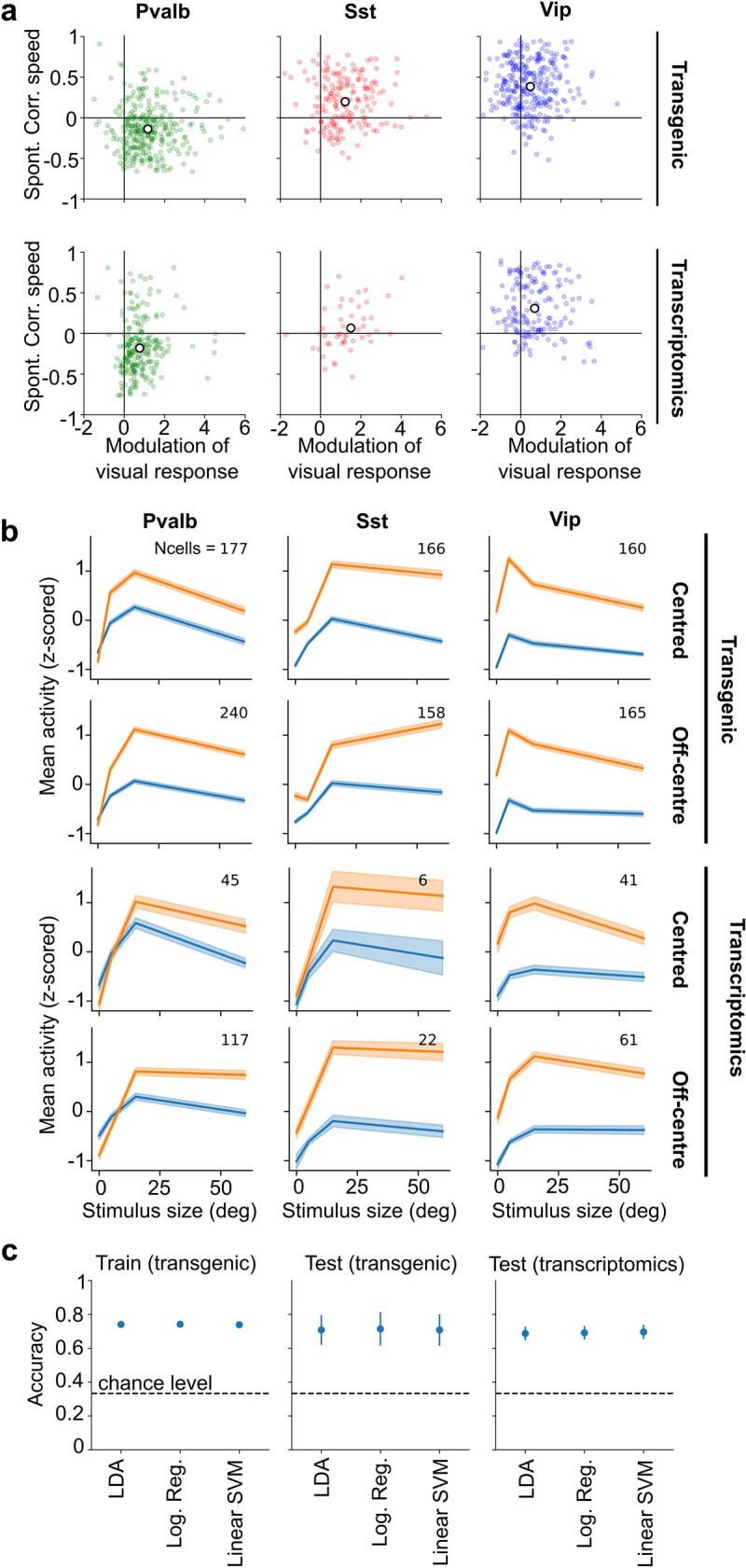
Transcriptomics has revealed that cortical inhibitory neurons exhibit a great diversity of fine molecular subtypes1-6, but it is not known whether these subtypes have correspondingly diverse patterns of activity in the living brain. Here we show that inhibitory subtypes in primary visual cortex (V1) have diverse correlates with brain state, which are organized by a single factor: position along the main axis of transcriptomic variation. We combined in vivo two-photon calcium imaging of mouse V1 with a transcriptomic method to identify mRNA for 72 selected genes in ex vivo slices. We classified inhibitory neurons imaged in layers 1-3 into a three-level hierarchy of 5 subclasses, 11 types and 35 subtypes using previously defined transcriptomic clusters3. Responses to visual stimuli differed significantly only between subclasses, with cells in the Sncg subclass uniformly suppressed, and cells in the other subclasses predominantly excited. Modulation by brain state differed at all hierarchical levels but could be largely predicted from the first transcriptomic principal component, which also predicted correlations with simultaneously recorded cells. Inhibitory subtypes that fired more in resting, oscillatory brain states had a smaller fraction of their axonal projections in layer 1, narrower spikes, lower input resistance and weaker adaptation as determined in vitro7, and expressed more inhibitory cholinergic receptors. Subtypes that fired more during arousal had the opposite properties. Thus, a simple principle may largely explain how diverse inhibitory V1 subtypes shape state-dependent cortical processing.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures














Comment in
-
A gene-expression axis defines neuron behaviour.Nature. 2022 Jul;607(7918):243-244. doi: 10.1038/d41586-022-01640-z. Nature. 2022. PMID: 35794379 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
