

This is a preprint.
SPLASH: a statistical, reference-free genomic algorithm unifies biological discovery
- PMID: 35794890
- PMCID: PMC9258296
- DOI: 10.1101/2022.06.24.497555
SPLASH: a statistical, reference-free genomic algorithm unifies biological discovery
Update in
-
SPLASH: A statistical, reference-free genomic algorithm unifies biological discovery.Cell. 2023 Dec 7;186(25):5440-5456.e26. doi: 10.1016/j.cell.2023.10.028. Cell. 2023. PMID: 38065078 Free PMC article.
Abstract
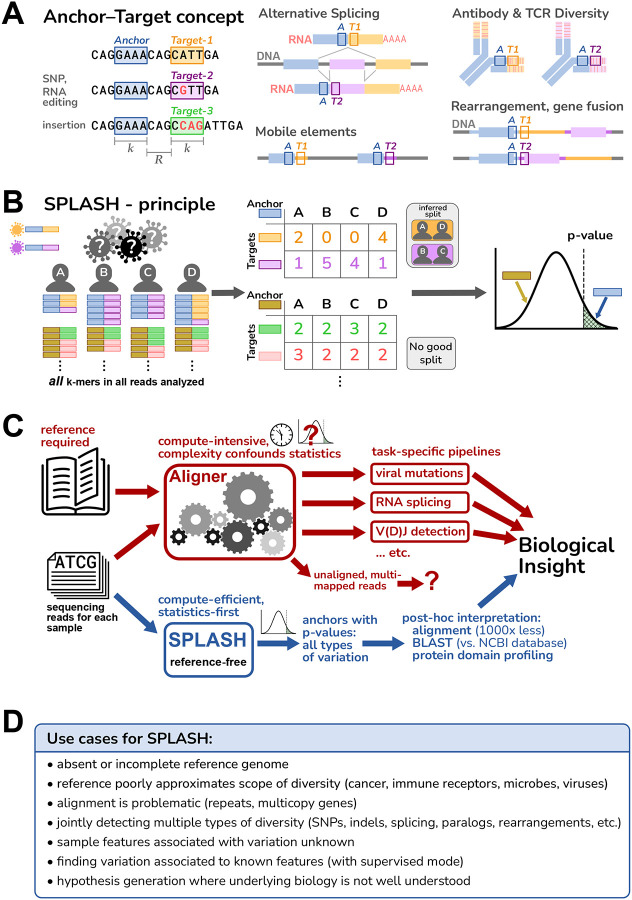
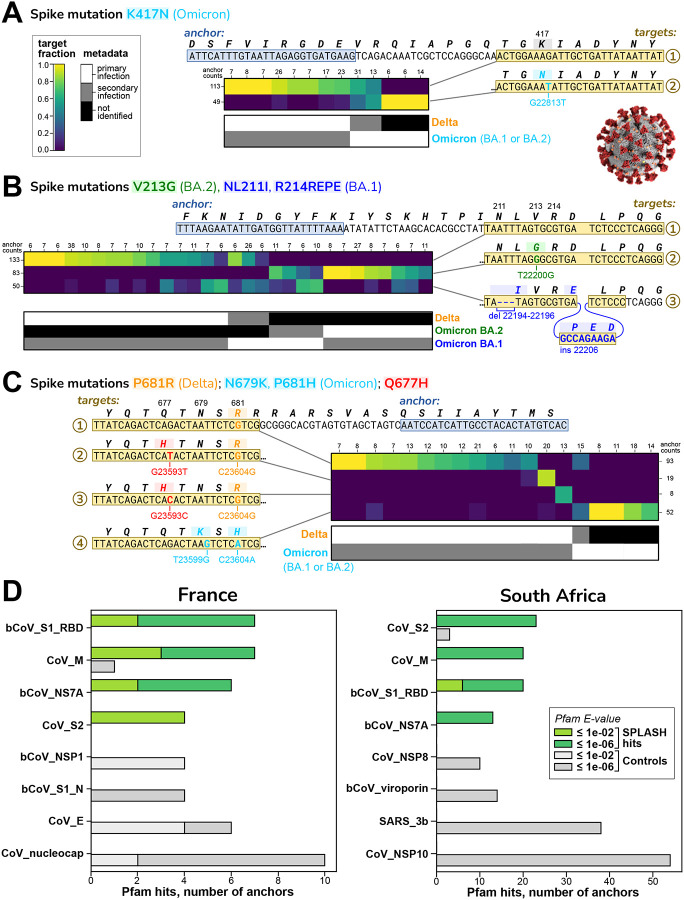
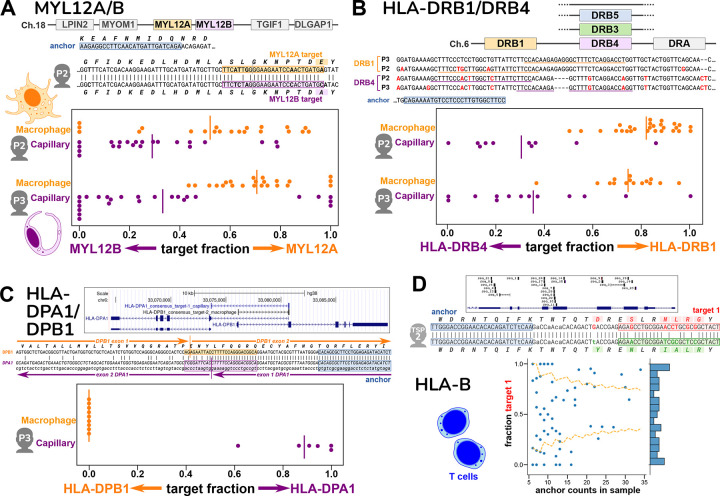
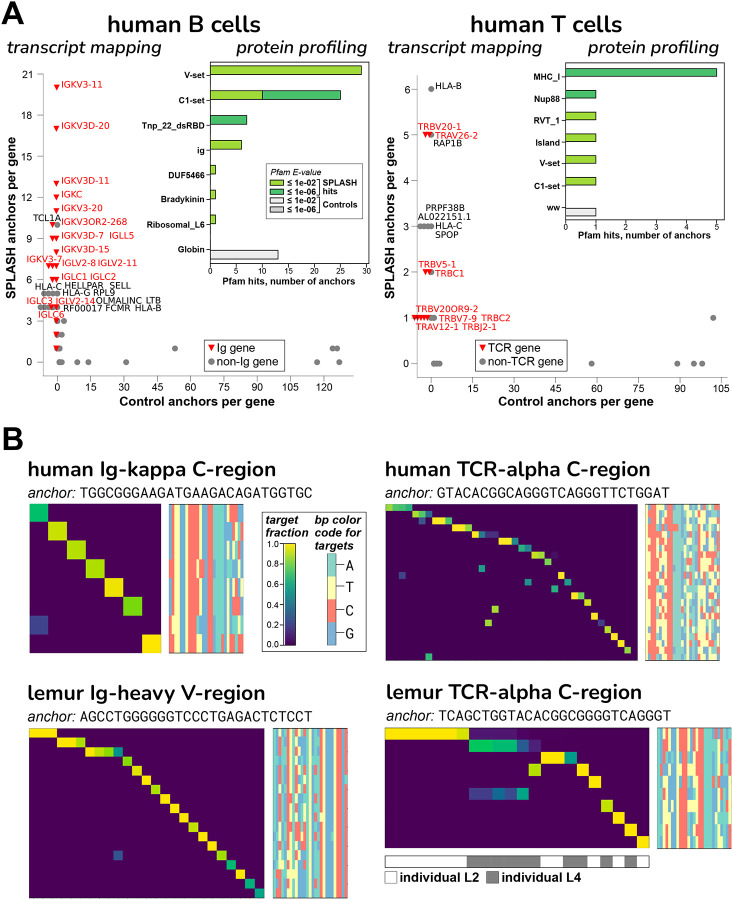
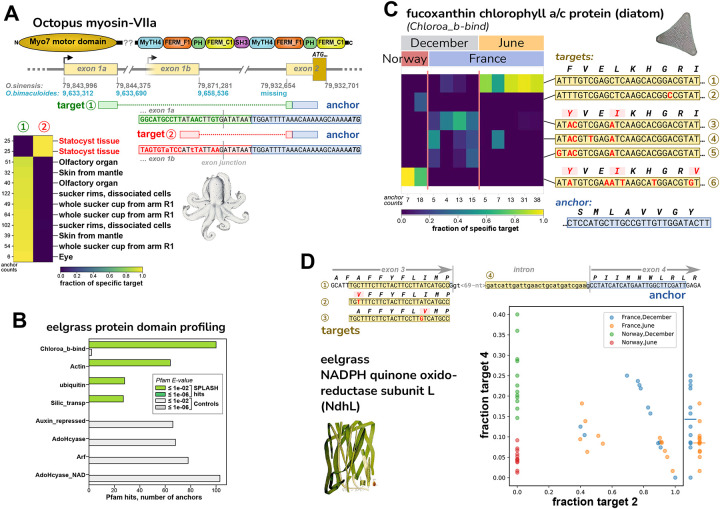
Today's genomics workflows typically require alignment to a reference sequence, which limits discovery. We introduce a new unifying paradigm, SPLASH (Statistically Primary aLignment Agnostic Sequence Homing), an approach that directly analyzes raw sequencing data to detect a signature of regulation: sample-specific sequence variation. The approach, which includes a new statistical test, is computationally efficient and can be run at scale. SPLASH unifies detection of myriad forms of sequence variation. We demonstrate that SPLASH identifies complex mutation patterns in SARS-CoV-2 strains, discovers regulated RNA isoforms at the single cell level, documents the vast sequence diversity of adaptive immune receptors, and uncovers biology in non-model organisms undocumented in their reference genomes: geographic and seasonal variation and diatom association in eelgrass, an oceanic plant impacted by climate change, and tissue-specific transcripts in octopus. SPLASH is a new unifying approach to genomic analysis that enables an expansive scope of discovery without metadata or references.
Conflict of interest statement
Competing interests K.C., T.Z.B. and J.S. are inventors on provisional patents related to this work. The authors declare no other competing interests.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous