

Physics-driven coarse-grained model for biomolecular phase separation with near-quantitative accuracy
- PMID: 35795820
- PMCID: PMC7612994
- DOI: 10.1038/s43588-021-00155-3
Physics-driven coarse-grained model for biomolecular phase separation with near-quantitative accuracy
Abstract
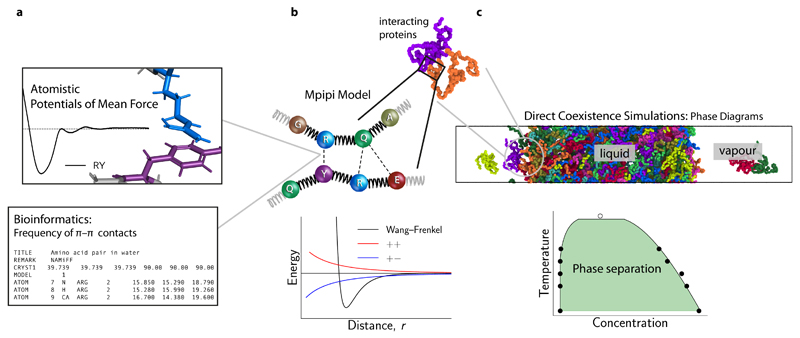
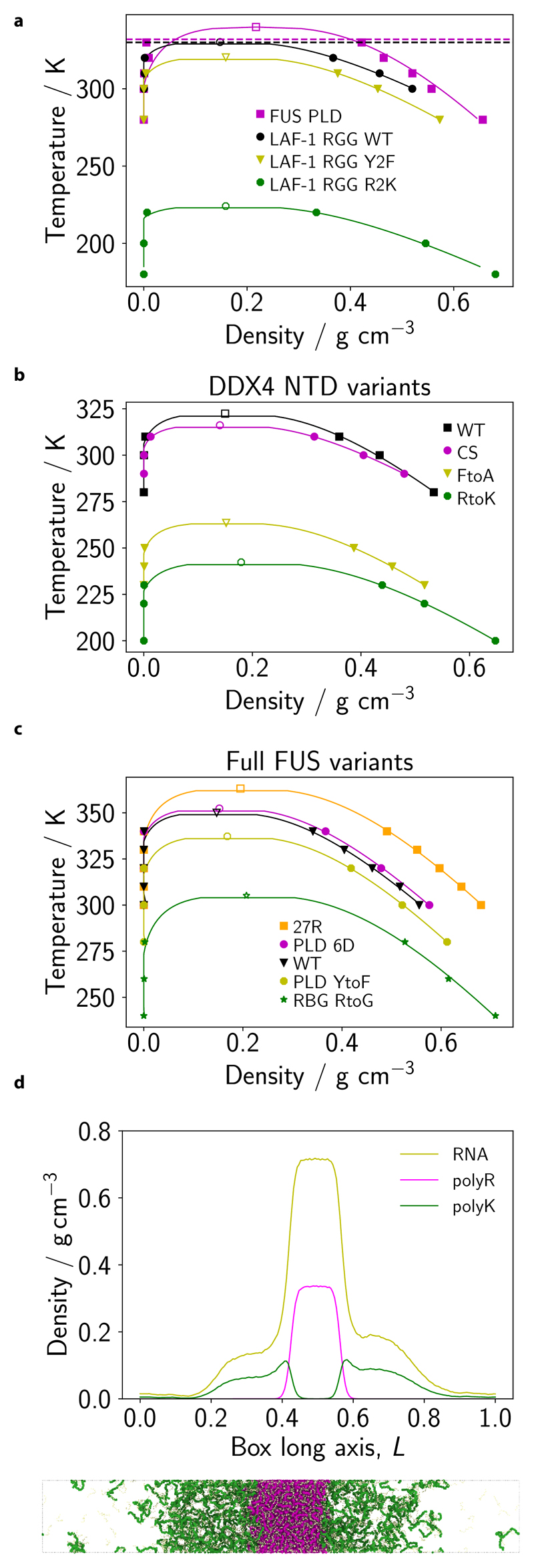
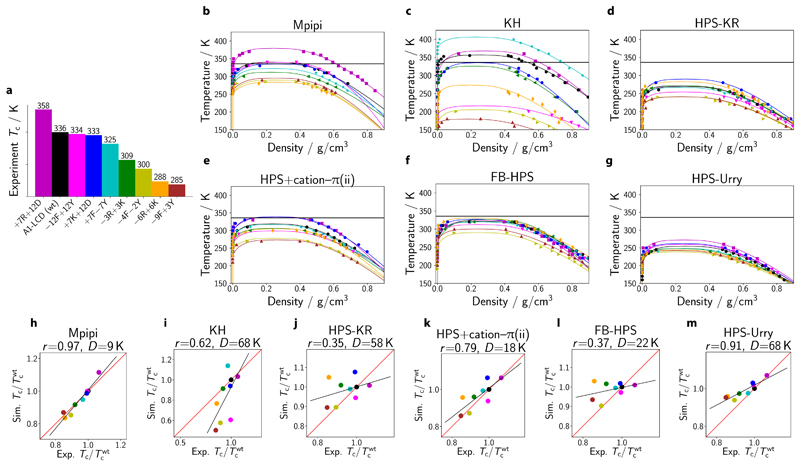
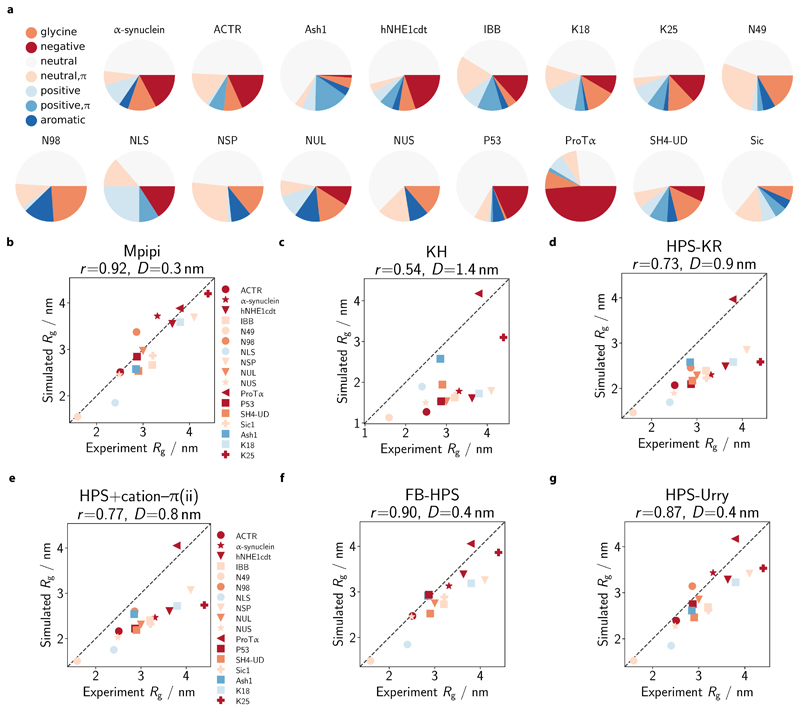
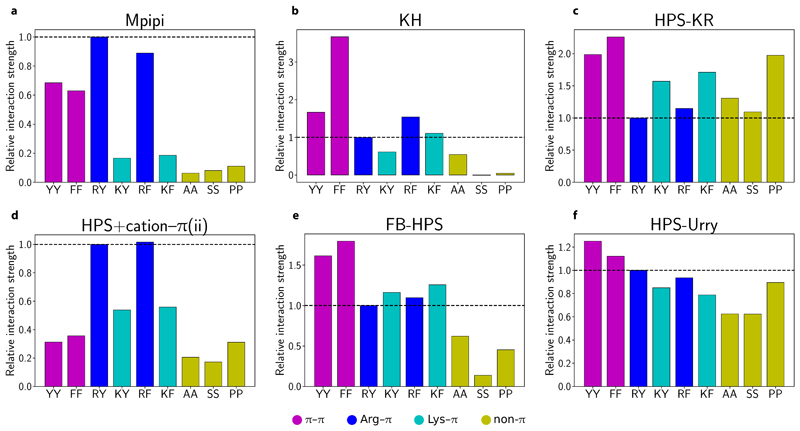
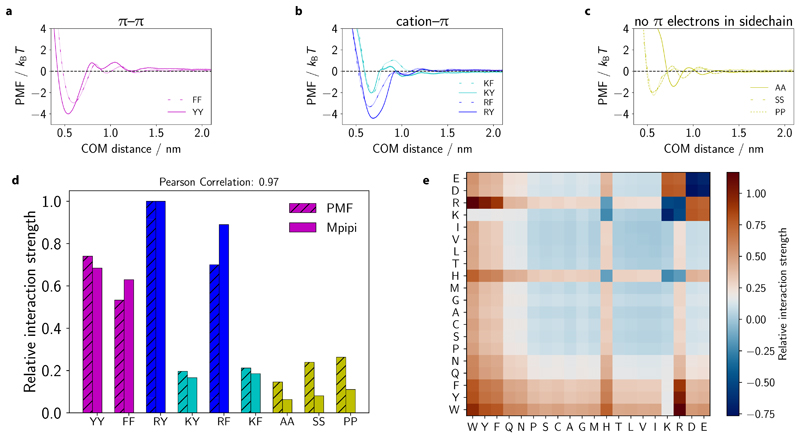
Various physics- and data-driven sequence-dependent protein coarse-grained models have been developed to study biomolecular phase separation and elucidate the dominant physicochemical driving forces. Here, we present Mpipi, a multiscale coarse-grained model that describes almost quantitatively the change in protein critical temperatures as a function of amino-acid sequence. The model is parameterised from both atomistic simulations and bioinformatics data and accounts for the dominant role of π-π and hybrid cation-π/π-π interactions and the much stronger attractive contacts established by arginines than lysines. We provide a comprehensive set of benchmarks for Mpipi and seven other residue-level coarse-grained models against experimental radii of gyration and quantitative in-vitro phase diagrams; Mpipi predictions agree well with experiment on both fronts. Moreover, it can account for protein-RNA interactions, correctly predicts the multiphase behaviour of a charge-matched poly-arginine/poly-lysine/RNA system, and recapitulates experimental LLPS trends for sequence mutations on FUS, DDX4 and LAF-1 proteins.
Conflict of interest statement
Competing Interests Statement The authors declare no competing interests.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
