

Regulation of TORC1 by MAPK Signaling Determines Sensitivity and Acquired Resistance to Trametinib in Pediatric BRAFV600E Brain Tumor Models
- PMID: 35797217
- PMCID: PMC10230442
- DOI: 10.1158/1078-0432.CCR-22-1052
Regulation of TORC1 by MAPK Signaling Determines Sensitivity and Acquired Resistance to Trametinib in Pediatric BRAFV600E Brain Tumor Models
Abstract
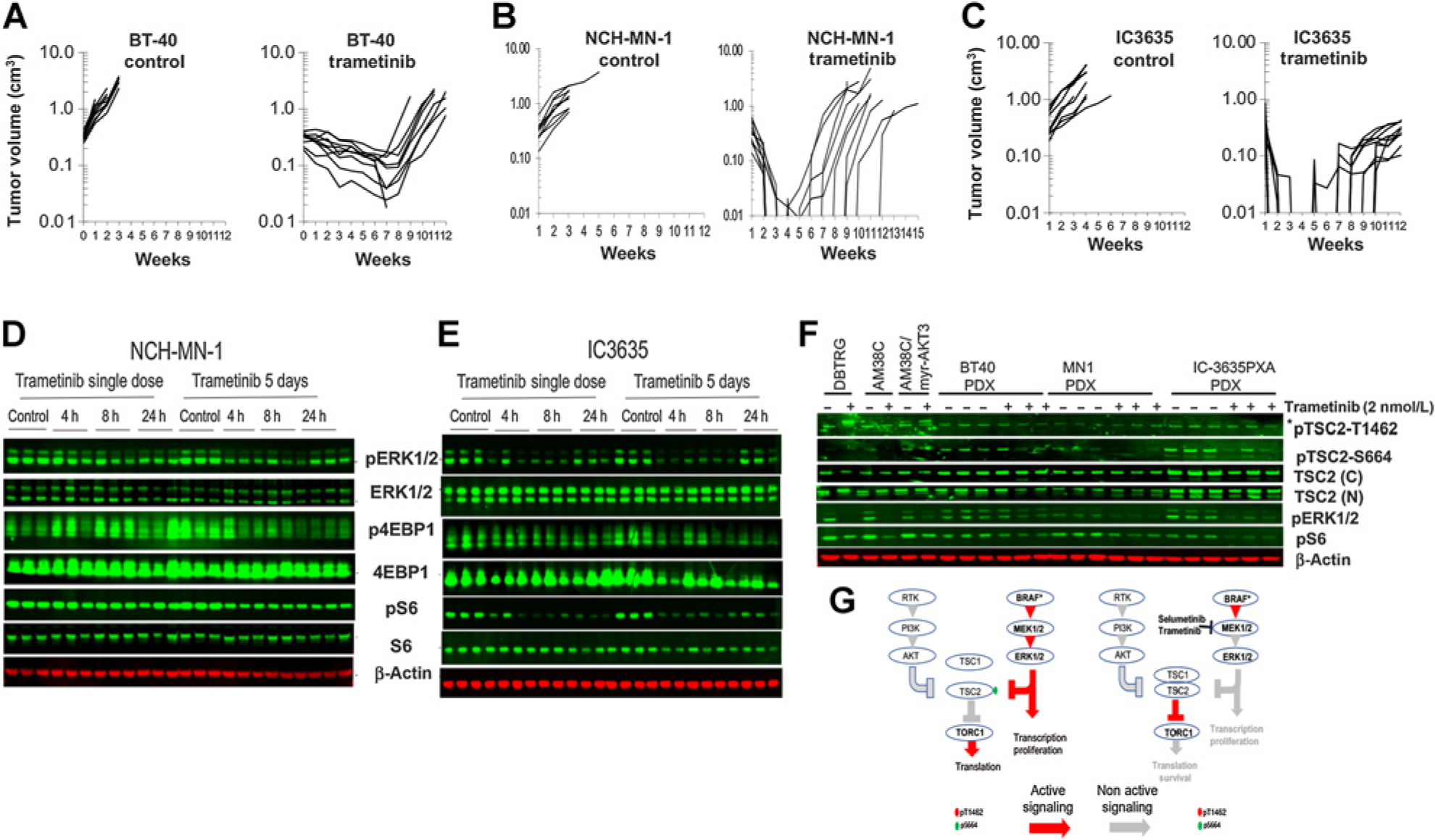
Purpose: We investigated why three patient-derived xenograft (PDX) childhood BRAFV600E-mutant brain tumor models are highly sensitive to trametinib. Mechanisms of acquired resistance selected in situ, and approaches to prevent resistance were also examined, which may translate to both low-grade glioma (LGG) molecular subtypes.
Experimental design: Sensitivity to trametinib [MEK inhibitor (MEKi)] alone or in combination with rapamycin (TORC1 inhibitor), was evaluated in pediatric PDX models. The effect of combined treatment of trametinib with rapamycin on development of trametinib resistance in vivo was examined. PDX tissue and tumor cells from trametinib-resistant xenografts were characterized.
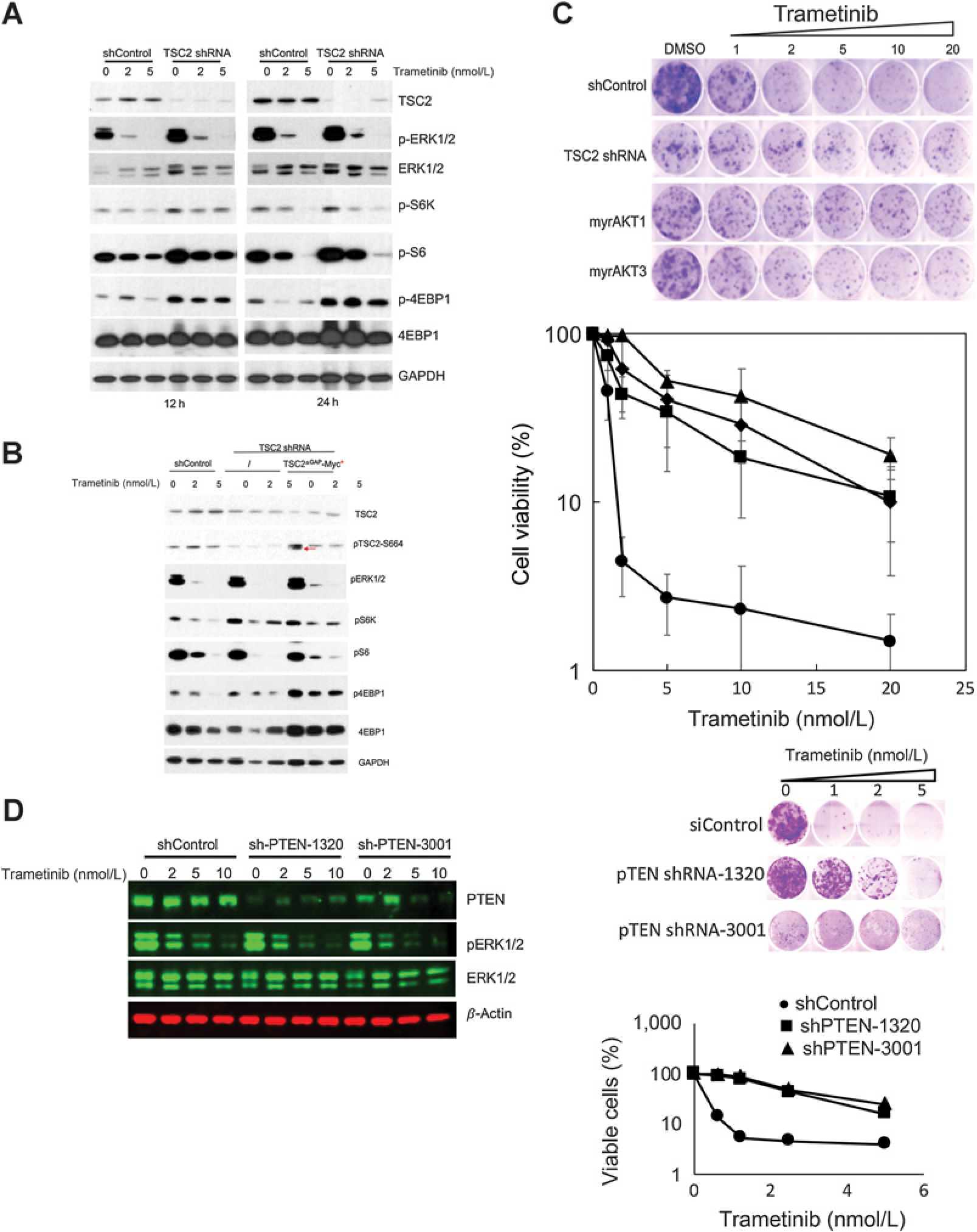
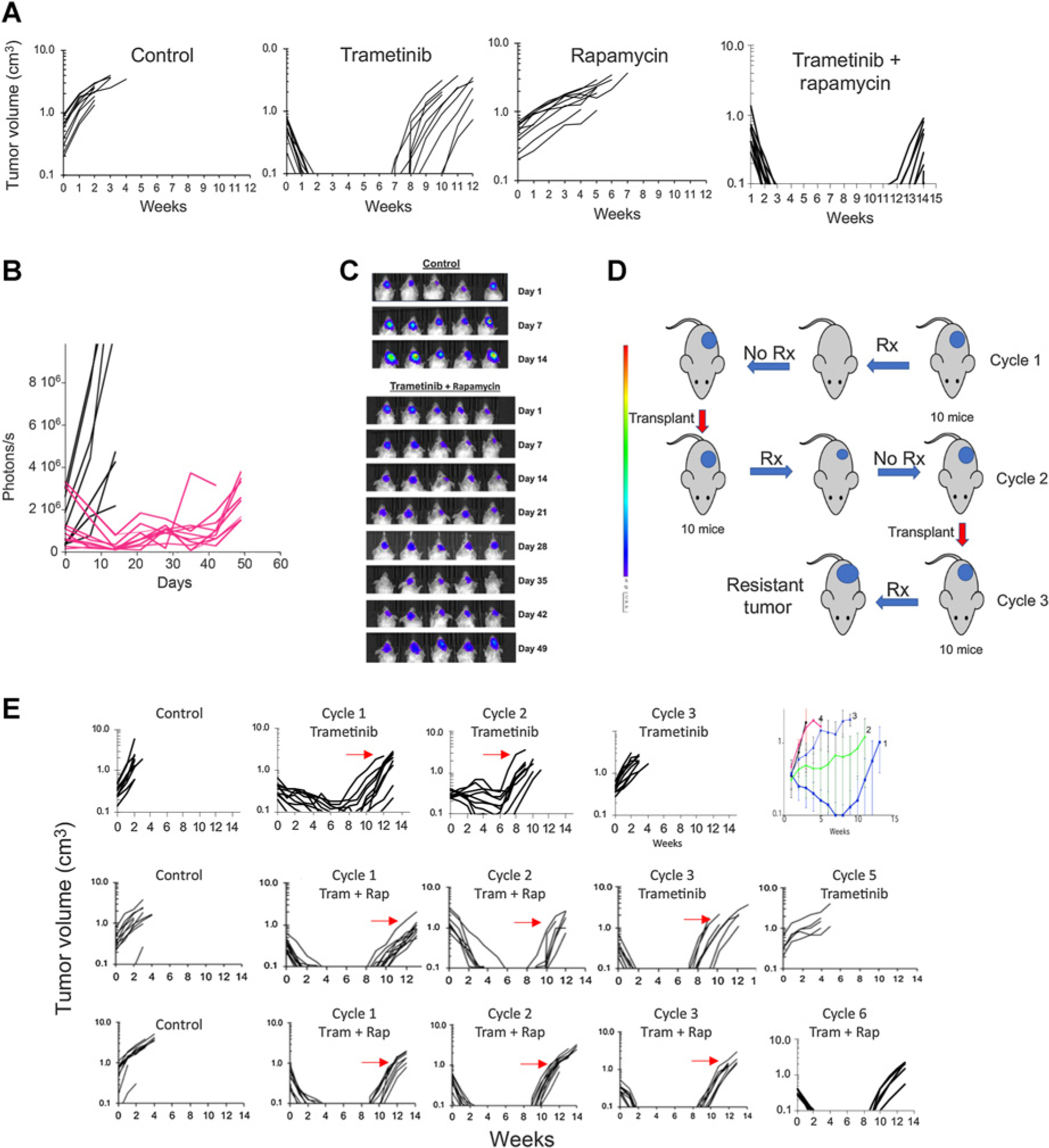
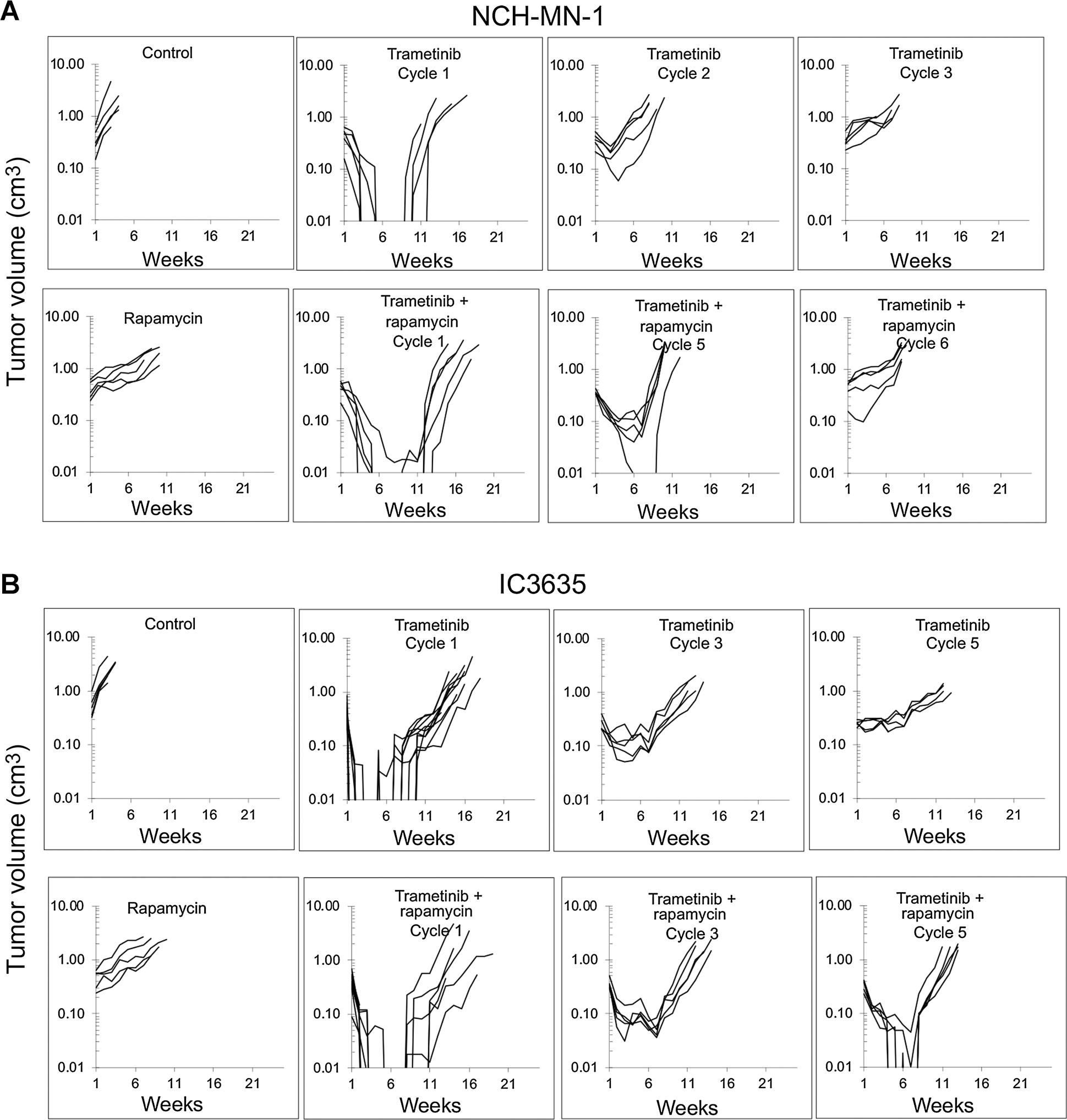
Results: In pediatric models TORC1 is activated through ERK-mediated inactivation of the tuberous sclerosis complex (TSC): consequently inhibition of MEK also suppressed TORC1 signaling. Trametinib-induced tumor regression correlated with dual inhibition of MAPK/TORC1 signaling, and decoupling TORC1 regulation from BRAF/MAPK control conferred trametinib resistance. In mice, acquired resistance to trametinib developed within three cycles of therapy in all three PDX models. Resistance to trametinib developed in situ is tumor-cell-intrinsic and the mechanism was tumor line specific. Rapamycin retarded or blocked development of resistance.
Conclusions: In these three pediatric BRAF-mutant brain tumors, TORC1 signaling is controlled by the MAPK cascade. Trametinib suppressed both MAPK/TORC1 pathways leading to tumor regression. While low-dose intermittent rapamycin to enhance inhibition of TORC1 only modestly enhanced the antitumor activity of trametinib, it prevented or retarded development of trametinib resistance, suggesting future therapeutic approaches using rapamycin analogs in combination with MEKis that may be therapeutically beneficial in both KIAA1549::BRAF- and BRAFV600E-driven gliomas.
©2022 American Association for Cancer Research.
Figures
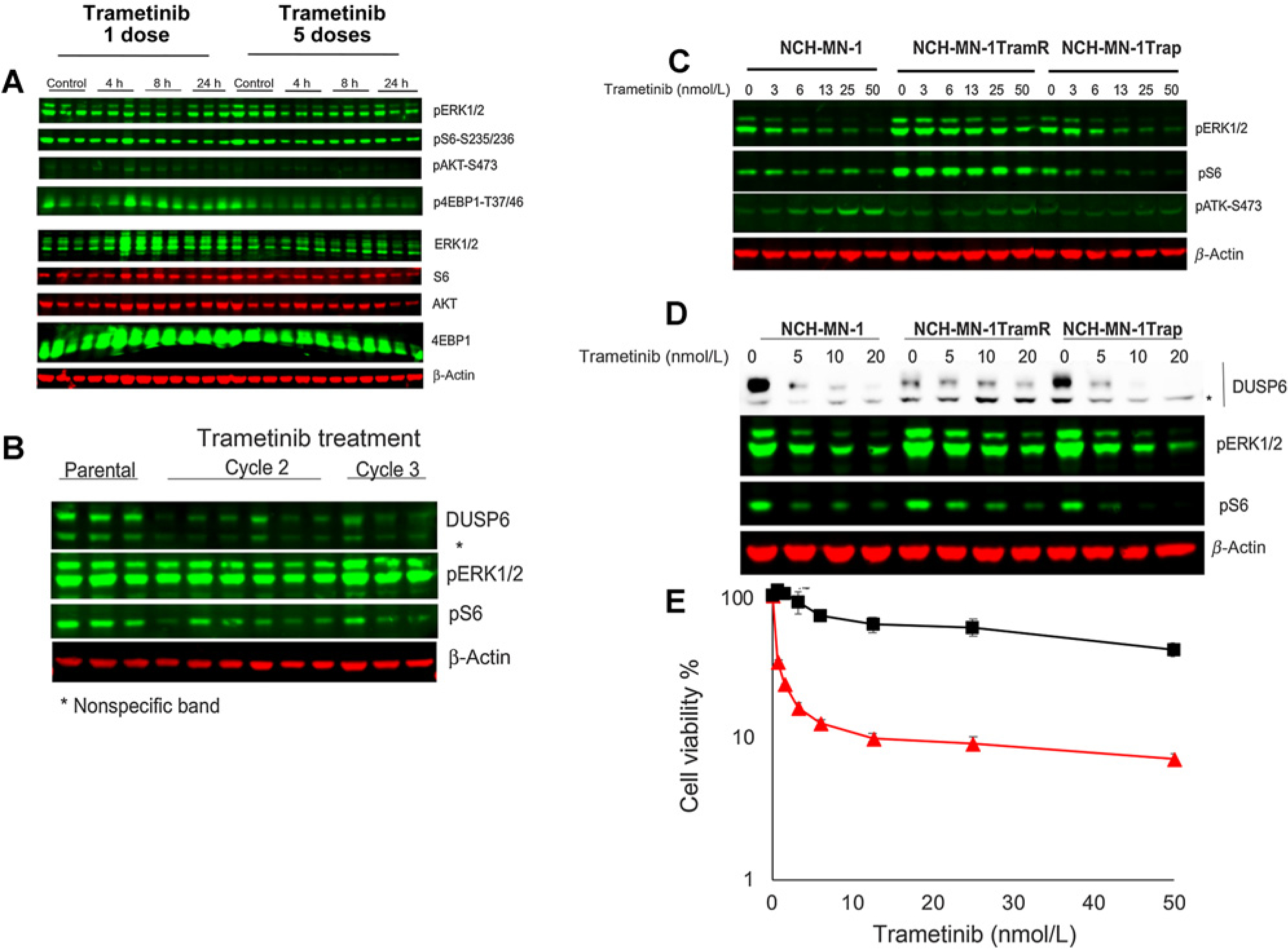
 red line) and trametinib-resistant xenografts (■ black line). Cells cultured in stem cell medium supplemented with FGF and rEGF were exposed to indicated concentration of trametinib for 72 hours. Cell proliferation was measured by Alamar Blue. h, hours.
red line) and trametinib-resistant xenografts (■ black line). Cells cultured in stem cell medium supplemented with FGF and rEGF were exposed to indicated concentration of trametinib for 72 hours. Cell proliferation was measured by Alamar Blue. h, hours.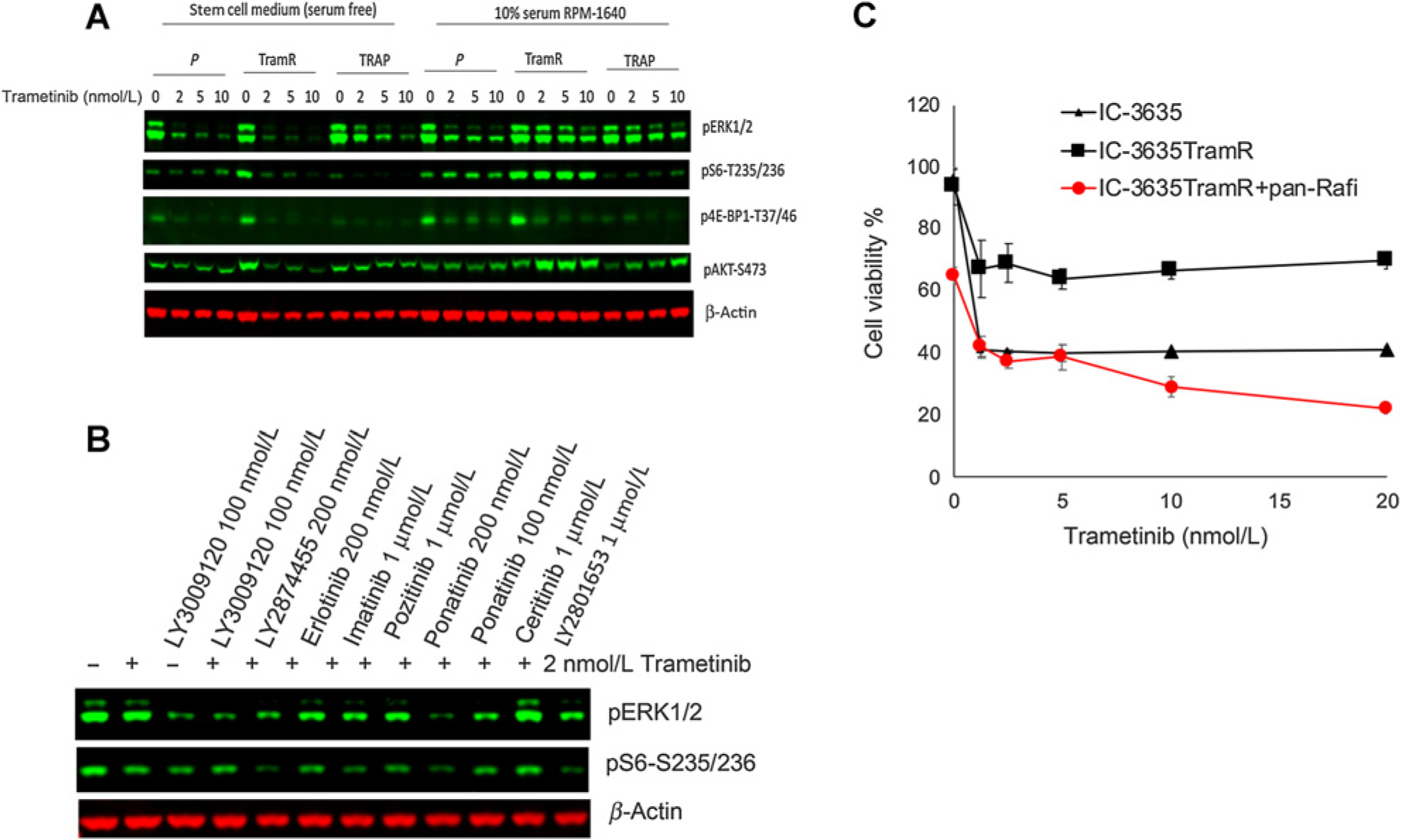
 ). Cells were cultured in stem cell medium supplemented with FGF and EGF and exposed to the indicated concentration of trametinib with or without combining panRaf inhibitor for 72 hours. Cell viability was measured by Alamar Blue, untreated cells were set as 100%.
). Cells were cultured in stem cell medium supplemented with FGF and EGF and exposed to the indicated concentration of trametinib with or without combining panRaf inhibitor for 72 hours. Cell viability was measured by Alamar Blue, untreated cells were set as 100%.References
-
- Gnekow AK, Falkenstein F, von Hornstein S, Zwiener I, Berkefeld S, Bison B, et al. Long-term follow-up of the multicenter, multidisciplinary treatment study HIT-LGG-1996 for low-grade glioma in children and adolescents of the german speaking society of pediatric oncology and hematology. Neuro Oncol 2012;14:1265–84. - PMC - PubMed
-
- Greenberger BA, Pulsifer MB, Ebb DH, MacDonald SM, Jones RM, Butler WE, et al. Clinical outcomes and late endocrine, neurocognitive, and visual profiles of proton radiation for pediatric low-grade gliomas. Int J Radiat Oncol Biol Phys 2014;89:1060–8. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
