

Cancer treatment-induced NAD+ depletion in premature senescence and late cardiovascular complications
- PMID: 35801078
- PMCID: PMC9258520
- DOI: 10.20517/jca.2022.13
Cancer treatment-induced NAD+ depletion in premature senescence and late cardiovascular complications
Abstract
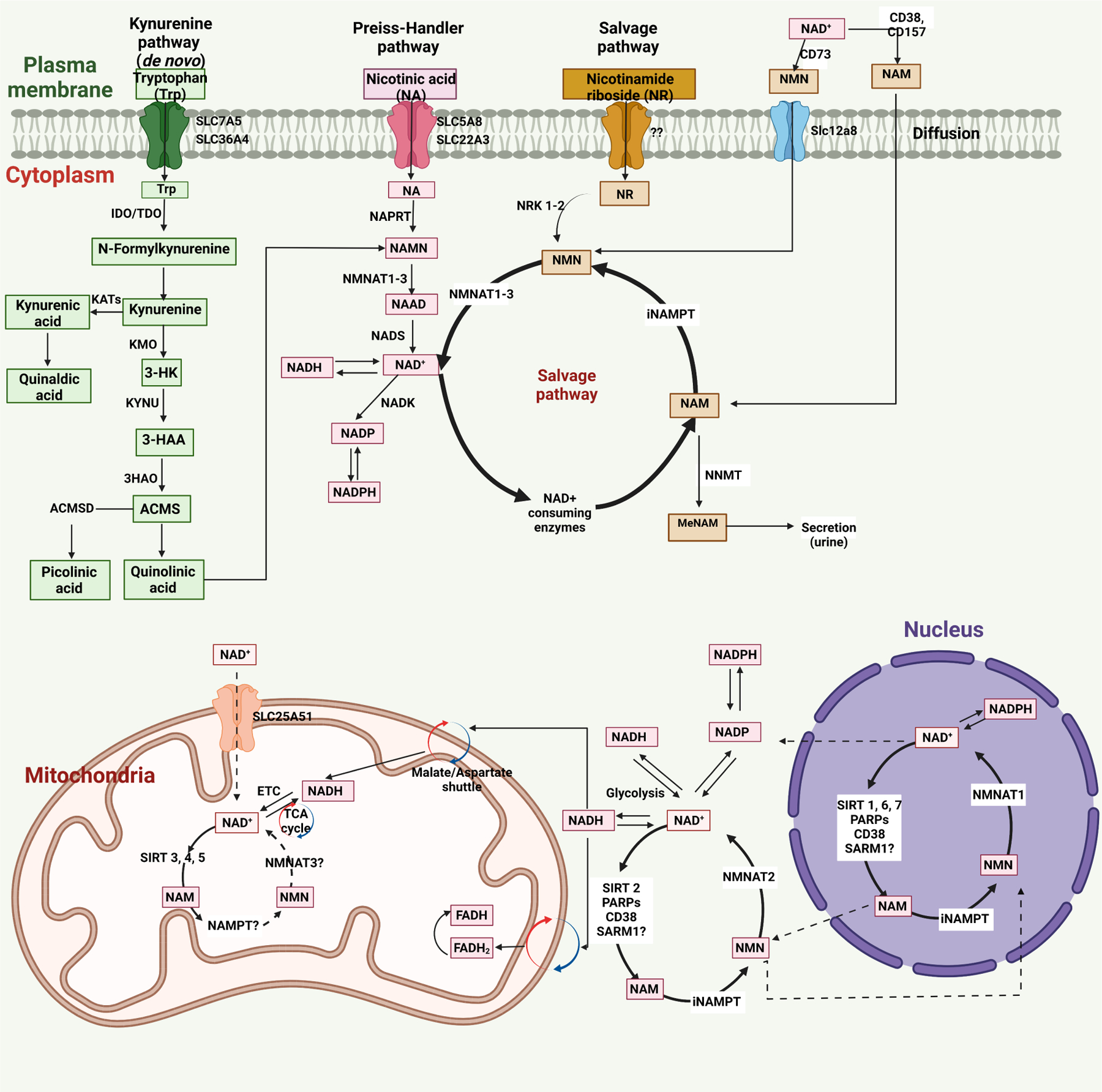
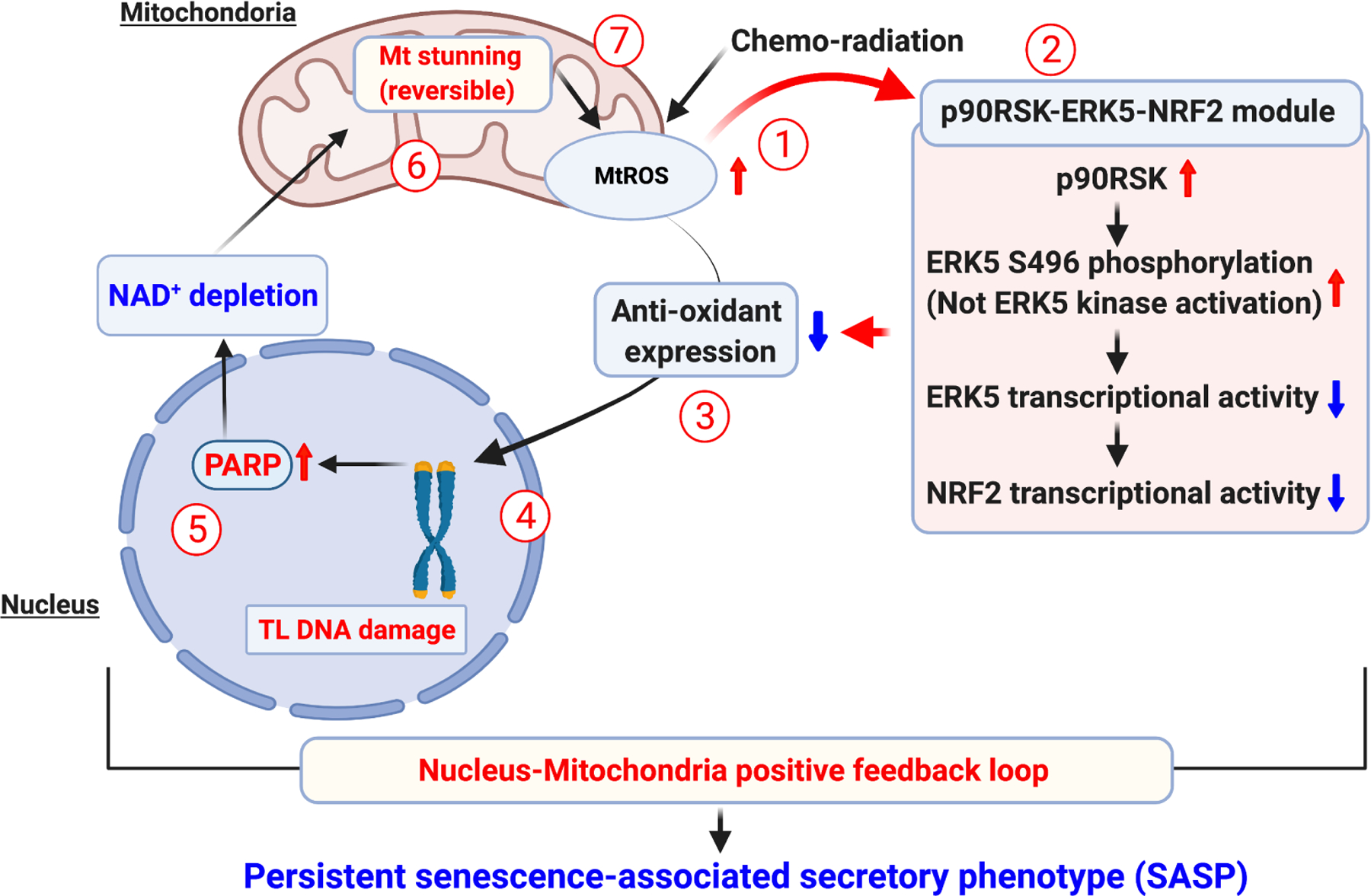
Numerous studies have revealed the critical role of premature senescence induced by various cancer treatment modalities in the pathogenesis of aging-related diseases. Senescence-associated secretory phenotype (SASP) can be induced by telomere dysfunction. Telomeric DNA damage response induced by some cancer treatments can persist for months, possibly accounting for long-term sequelae of cancer treatments. Telomeric DNA damage-induced mitochondrial dysfunction and increased reactive oxygen species production are hallmarks of premature senescence. Recently, we reported that the nucleus-mitochondria positive feedback loop formed by p90 ribosomal S6 kinase (p90RSK) and phosphorylation of S496 on ERK5 (a unique member of the mitogen-activated protein kinase family that is not only a kinase but also a transcriptional co-activator) were vital signaling events that played crucial roles in linking mitochondrial dysfunction, nuclear telomere dysfunction, persistent SASP induction, and atherosclerosis. In this review, we will discuss the role of NAD+ depletion in instigating SASP and its downstream signaling and regulatory mechanisms that lead to the premature onset of atherosclerotic cardiovascular diseases in cancer survivors.
Keywords: ERK5; NAD+; cardiovascular diseases; p90RSK; senescence-associated secretory phenotype (SASP).
Conflict of interest statement
Conflicts of interest All authors declare that there are no conflicts of interest.
Figures
References
-
- Syrigos KN, Karachalios D, Karapanagiotou EM, Nutting CM, Manolopoulos L, Harrington KJ. Head and neck cancer in the elderly: an overview on the treatment modalities. Cancer Treat Rev 2009;35:237–45. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous