

Genome-wide association study in quinoa reveals selection pattern typical for crops with a short breeding history
- PMID: 35801689
- PMCID: PMC9388097
- DOI: 10.7554/eLife.66873
Genome-wide association study in quinoa reveals selection pattern typical for crops with a short breeding history
Abstract
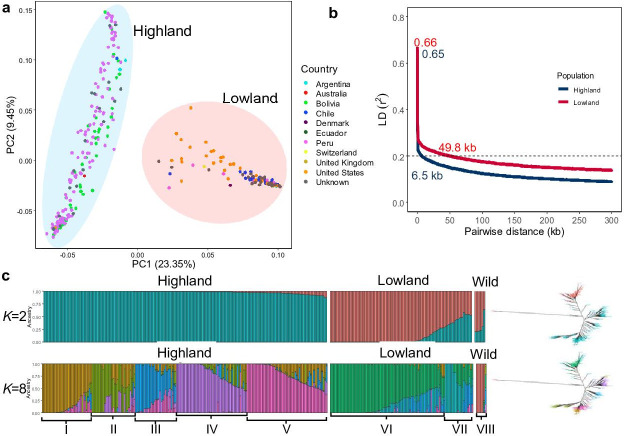
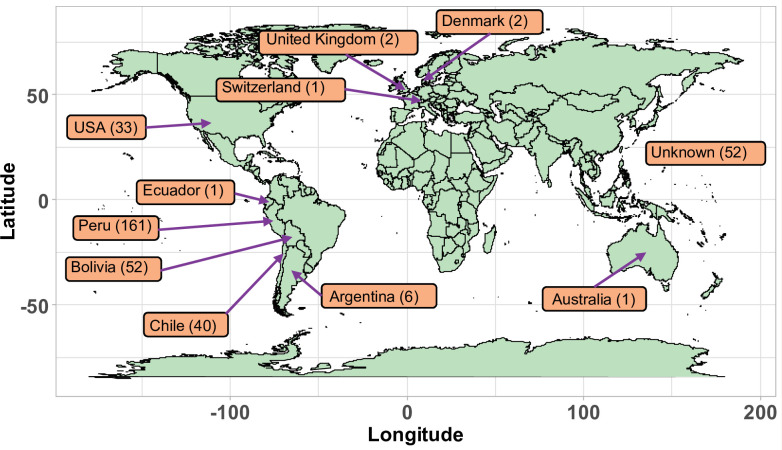
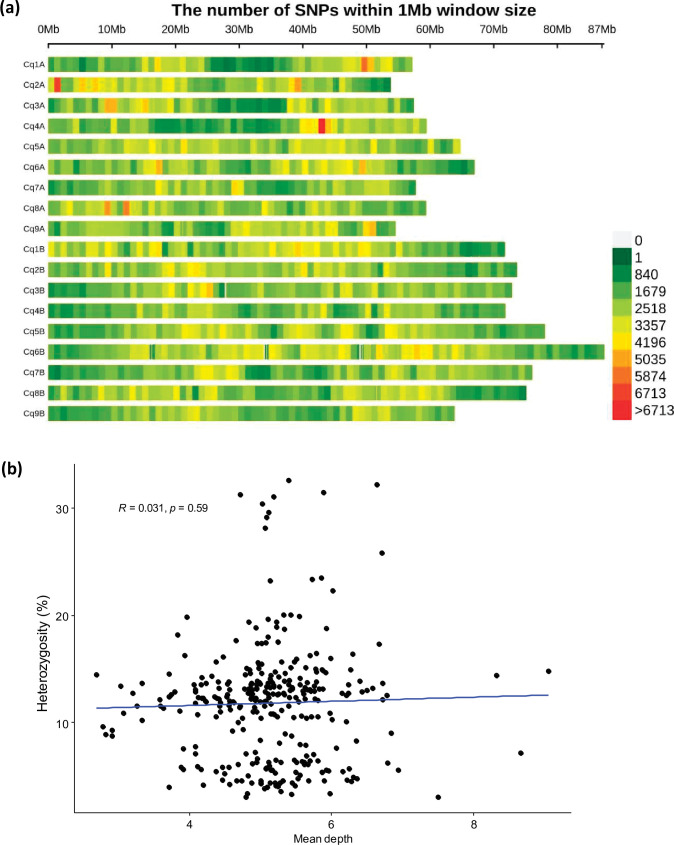
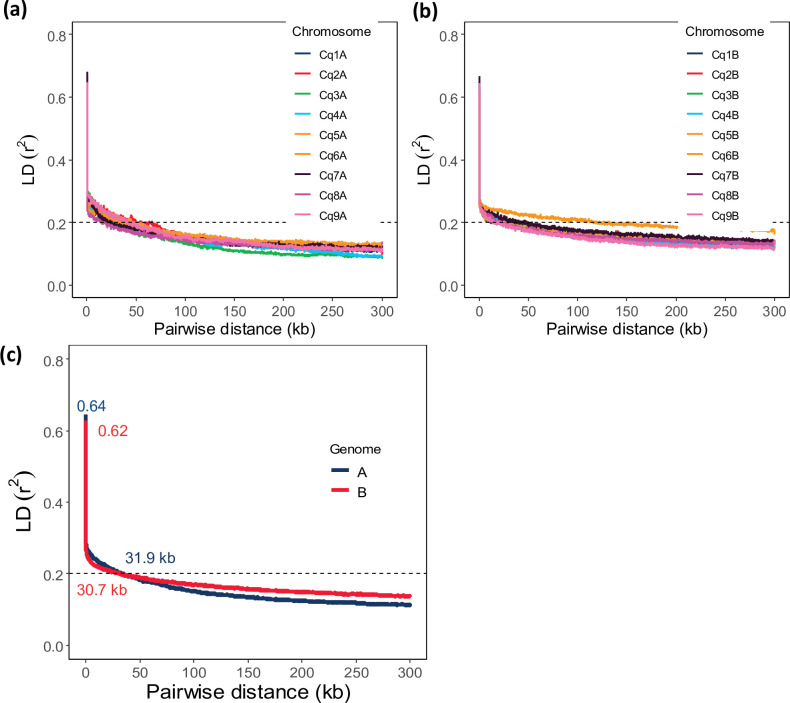
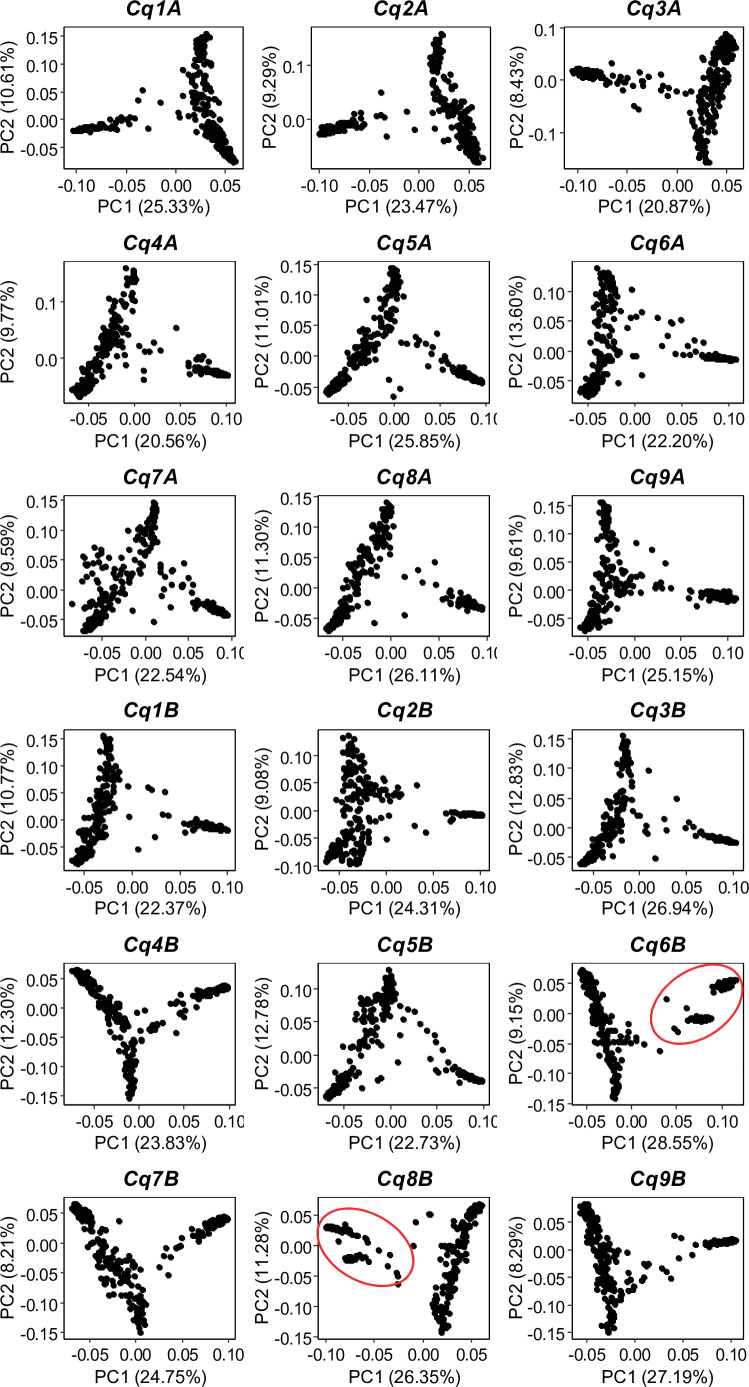
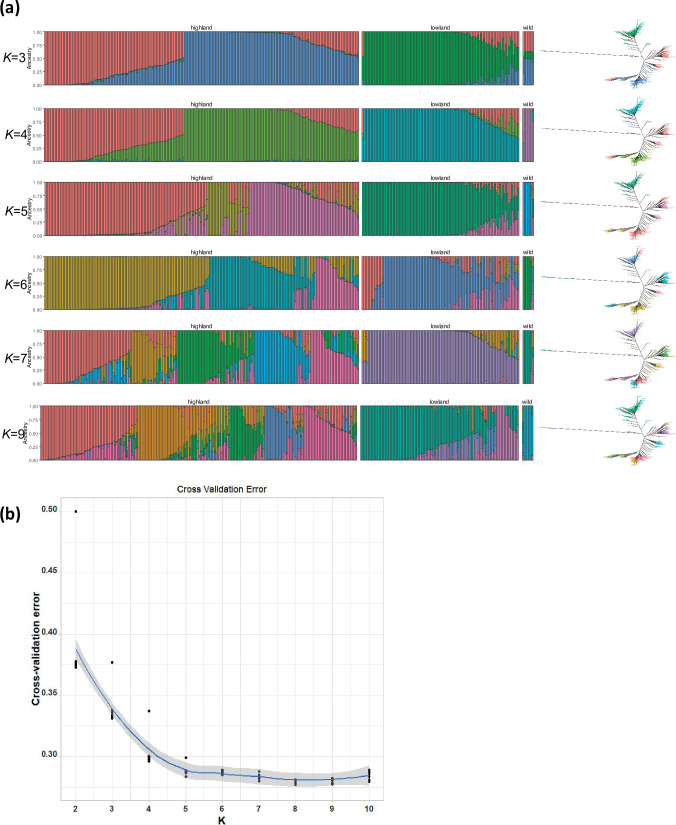
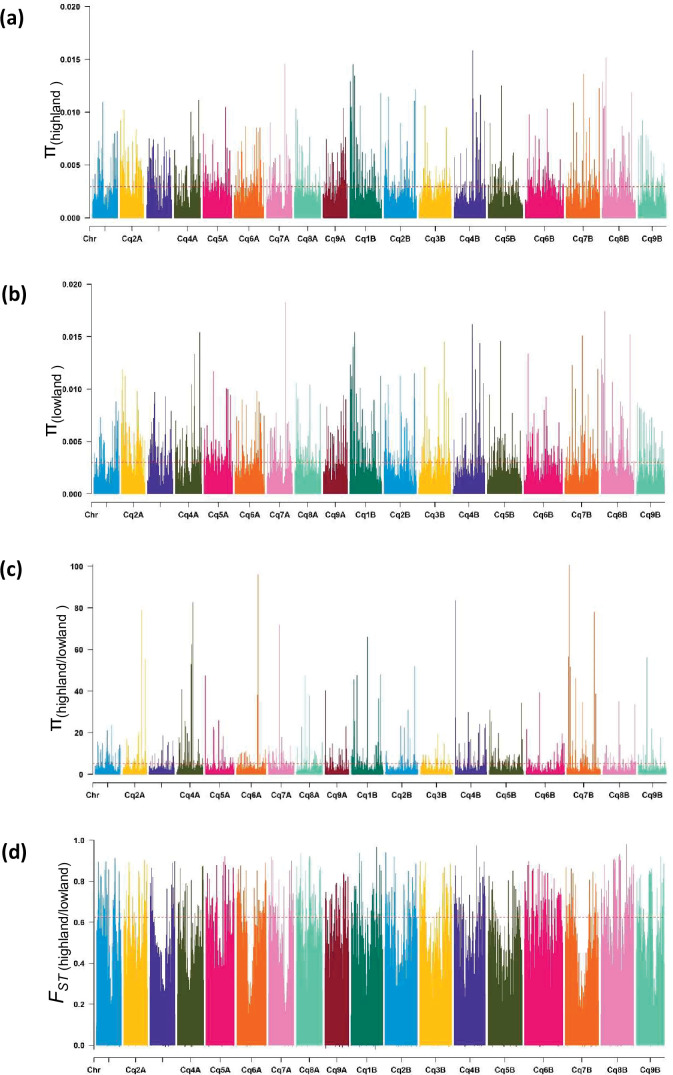
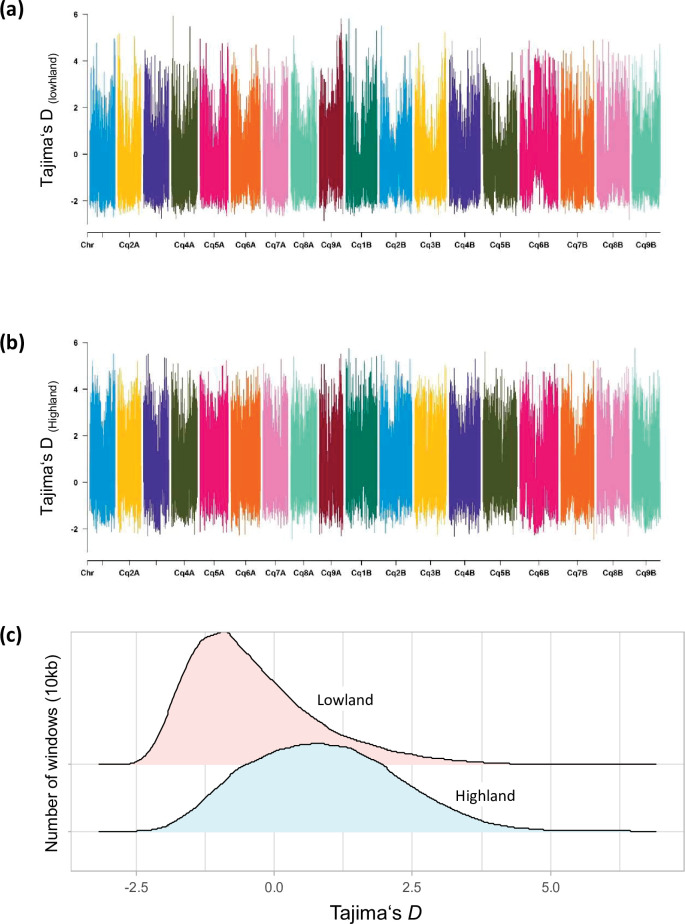
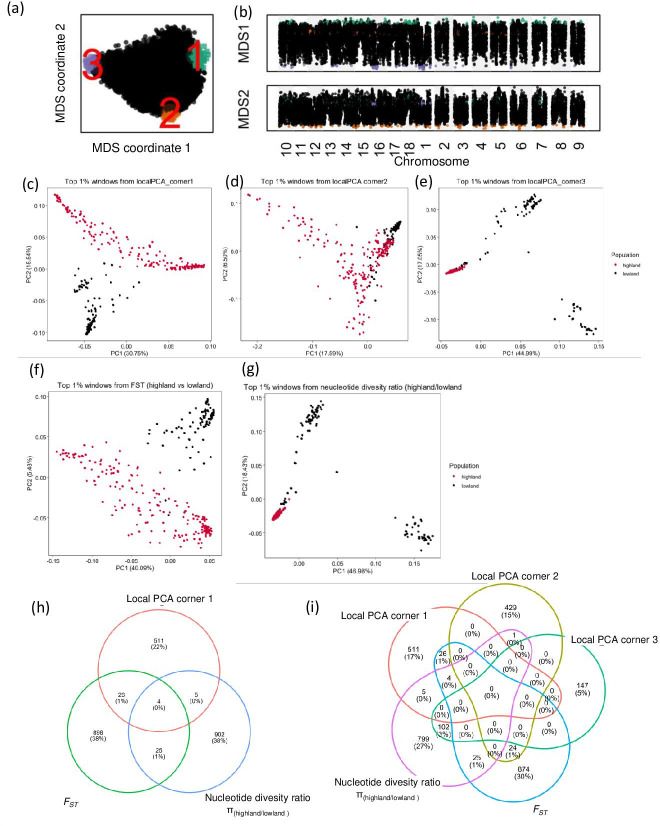
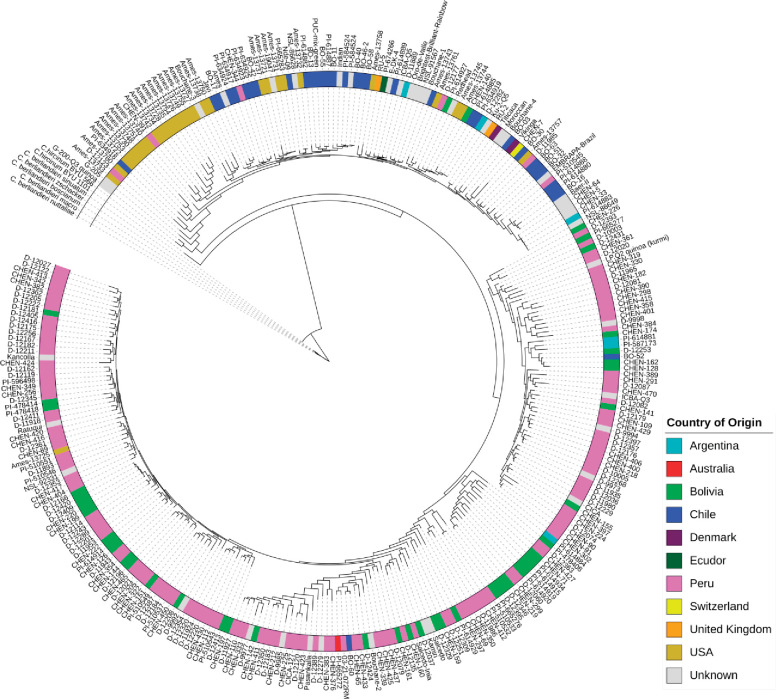
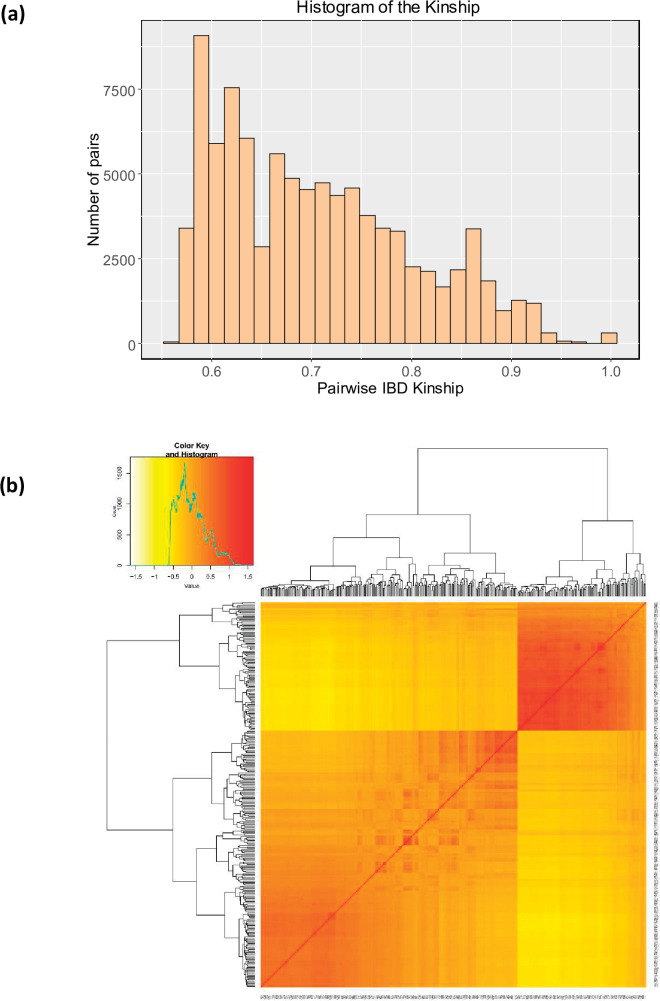
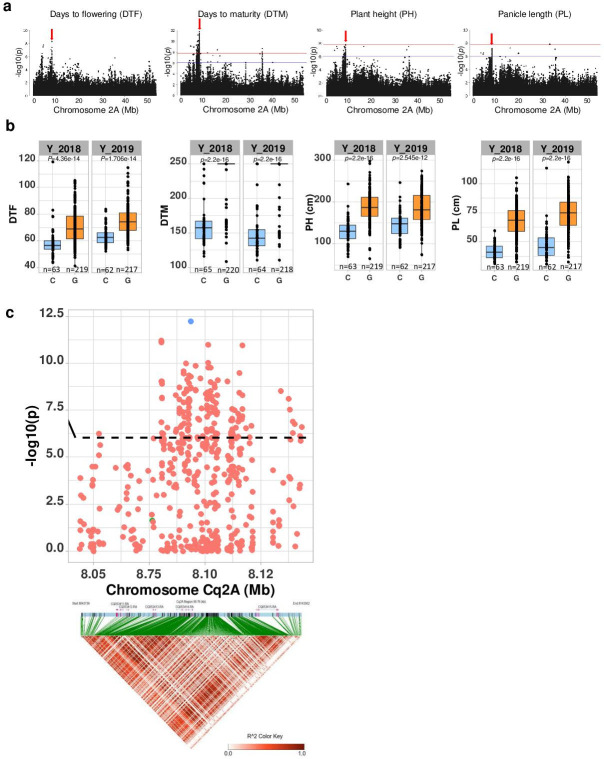
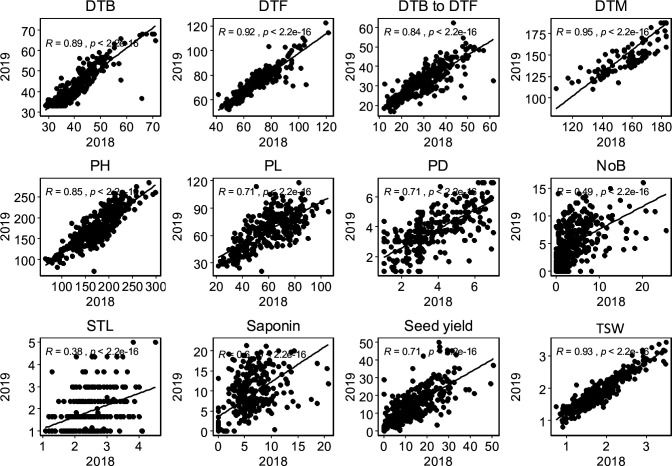
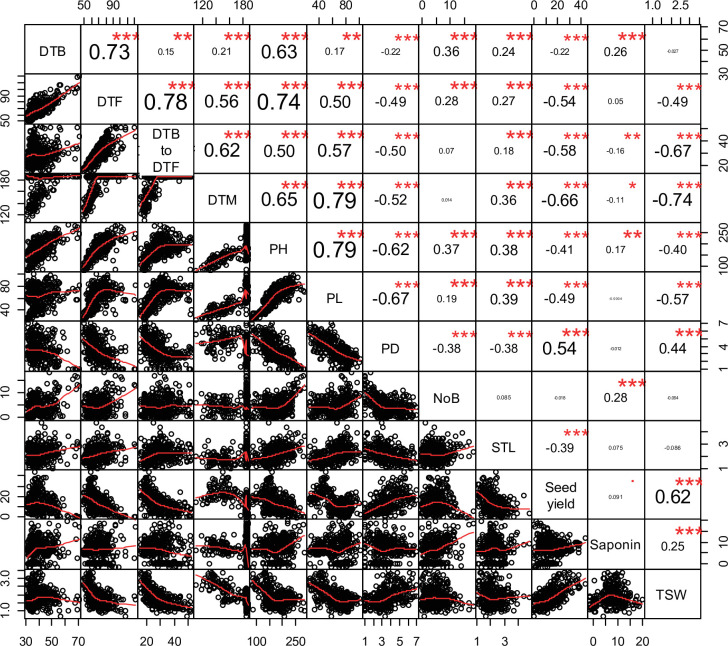
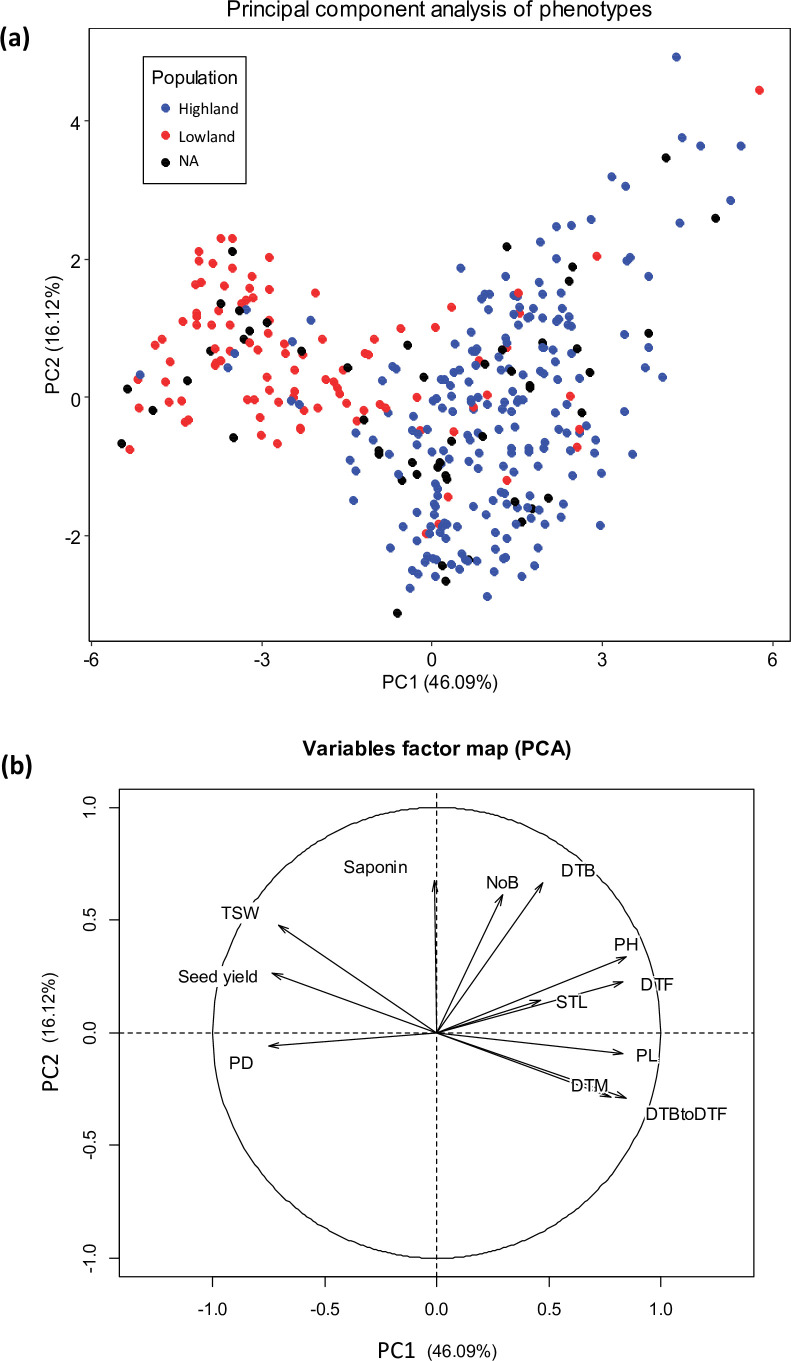
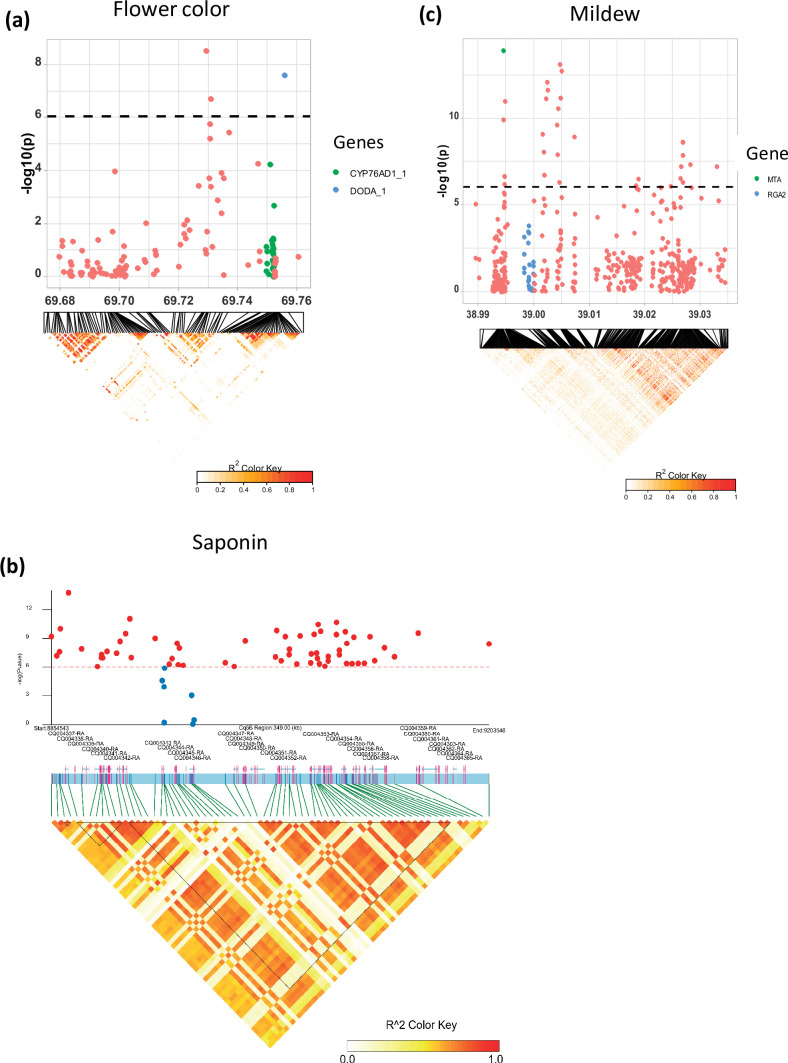
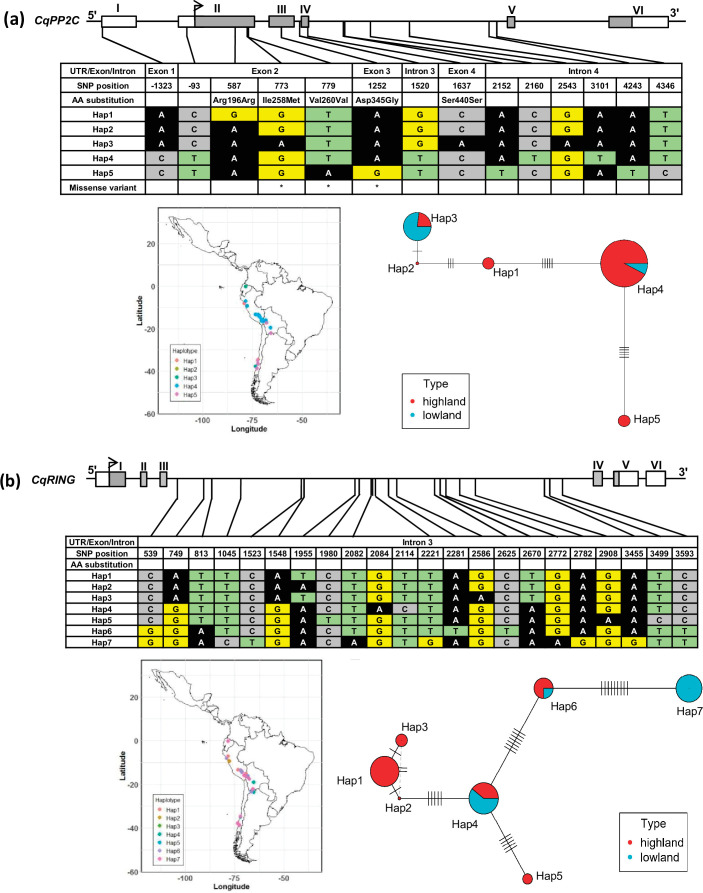
Quinoa germplasm preserves useful and substantial genetic variation, yet it remains untapped due to a lack of implementation of modern breeding tools. We have integrated field and sequence data to characterize a large diversity panel of quinoa. Whole-genome sequencing of 310 accessions revealed 2.9 million polymorphic high confidence single nucleotide polymorphism (SNP) loci. Highland and Lowland quinoa were clustered into two main groups, with FST divergence of 0.36 and linkage disequilibrium (LD) decay of 6.5 and 49.8 kb, respectively. A genome-wide association study using multi-year phenotyping trials uncovered 600 SNPs stably associated with 17 traits. Two candidate genes are associated with thousand seed weight, and a resistance gene analog is associated with downy mildew resistance. We also identified pleiotropically acting loci for four agronomic traits important for adaptation. This work demonstrates the use of re-sequencing data of an orphan crop, which is partially domesticated to rapidly identify marker-trait association and provides the underpinning elements for genomics-enabled quinoa breeding.
Keywords: Chenopodium quinoa; adaptation; domestication; genetic variation; genetics; genomics; plant breeding; re-sequencing.
Plain language summary
As human populations grow and climate change tightens its grip, developing nutritious crops which can thrive on poor soil and under difficult conditions will become a priority. Quinoa, a harvest currently overlooked by agricultural research, could be an interesting candidate in this effort. With its high nutritional value and its ability to tolerate drought, frost and high concentrations of salt in the soil, this hardy crop has been cultivated in the Andes for the last 5,000 to 7,000 years. Today its commercial production is mainly limited to Peru, Bolivia, and Ecuador. Pinpointing the genetic regions that control traits such as yields or flowering time would help agronomists to create new varieties better suited to life under northern latitudes and mechanical farming. To identify these genes, Patiranage et al. grew 310 varieties of quinoa from all over the world under the same conditions; the genomes of these plants were also examined in great detail. Analyses were then performed to link specific genetic variations with traits relevant to agriculture, helping to pinpoint changes in the genetic code linked to differences in how the plants grew, resisted disease, or produced seeds of varying quality. Candidate genes likely to control these traits were then put forward. The study by Patiranage et al. provides a genetic map where genes of agronomical importance have been precisely located and their effects measured. This resource will help to select genetic profiles which could be used to create new quinoa breeds better adapted to a changing world.
© 2022, Patiranage et al.
Conflict of interest statement
DP, ER, NE, GW, KS, SS, MT, CJ No competing interests declared
Figures

Similar articles
-
Mining genomic regions associated with agronomic and biochemical traits in quinoa through GWAS.Sci Rep. 2024 Apr 22;14(1):9205. doi: 10.1038/s41598-024-59565-8. Sci Rep. 2024. PMID: 38649738 Free PMC article.
-
Genomic Variation Underlying the Breeding Selection of Quinoa Varieties Longli-4 and CA3-1 in China.Int J Mol Sci. 2022 Nov 14;23(22):14030. doi: 10.3390/ijms232214030. Int J Mol Sci. 2022. PMID: 36430511 Free PMC article.
-
Developing Chenopodium ficifolium as a potential B genome diploid model system for genetic characterization and improvement of allotetraploid quinoa (Chenopodium quinoa).BMC Plant Biol. 2021 Oct 25;21(1):490. doi: 10.1186/s12870-021-03270-5. BMC Plant Biol. 2021. PMID: 34696717 Free PMC article.
-
Progress on genomics and locus of important agronomic traits in Chenopodium quinoa.Yi Chuan. 2022 Nov 20;44(11):1009-1027. doi: 10.16288/j.yczz.22-289. Yi Chuan. 2022. PMID: 36384994 Review.
-
Understanding and utilizing crop genome diversity via high-resolution genotyping.Plant Biotechnol J. 2016 Apr;14(4):1086-94. doi: 10.1111/pbi.12456. Epub 2015 Aug 19. Plant Biotechnol J. 2016. PMID: 27003869 Free PMC article. Review.
Cited by
-
Quinoa: A Promising Crop for Resolving the Bottleneck of Cultivation in Soils Affected by Multiple Environmental Abiotic Stresses.Plants (Basel). 2024 Jul 31;13(15):2117. doi: 10.3390/plants13152117. Plants (Basel). 2024. PMID: 39124236 Free PMC article. Review.
-
Empirical phenotyping and genome-wide association study reveal the association of panicle architecture with yield in Chenopodium quinoa.Front Microbiol. 2024 Mar 18;15:1349239. doi: 10.3389/fmicb.2024.1349239. eCollection 2024. Front Microbiol. 2024. PMID: 38562468 Free PMC article.
-
A comprehensive characterization of agronomic and end-use quality phenotypes across a quinoa world core collection.Front Plant Sci. 2023 Feb 16;14:1101547. doi: 10.3389/fpls.2023.1101547. eCollection 2023. Front Plant Sci. 2023. PMID: 36875583 Free PMC article.
-
High-Density Mapping of Quantitative Trait Loci Controlling Agronomically Important Traits in Quinoa (Chenopodium quinoa Willd.).Front Plant Sci. 2022 Jun 9;13:916067. doi: 10.3389/fpls.2022.916067. eCollection 2022. Front Plant Sci. 2022. PMID: 35812962 Free PMC article.
-
Root restriction accelerates genomic target identification in quinoa under controlled conditions.Physiol Plant. 2025 Mar-Apr;177(2):e70223. doi: 10.1111/ppl.70223. Physiol Plant. 2025. PMID: 40231839 Free PMC article.
References
-
- Abrouk M, Ahmed HI, Cubry P, Šimoníková D, Cauet S, Pailles Y, Bettgenhaeuser J, Gapa L, Scarcelli N, Couderc M, Zekraoui L, Kathiresan N, Čížková J, Hřibová E, Doležel J, Arribat S, Bergès H, Wieringa JJ, Gueye M, Kane NA, Leclerc C, Causse S, Vancoppenolle S, Billot C, Wicker T, Vigouroux Y, Barnaud A, Krattinger SG. Fonio millet genome unlocks African orphan crop diversity for agriculture in a changing climate. Nature Communications. 2020;11:4488. doi: 10.1038/s41467-020-18329-4. - DOI - PMC - PubMed
-
- Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Soft. 2015;67:48. doi: 10.18637/jss.v067.i01. - DOI
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
