

Determination of human DNA replication origin position and efficiency reveals principles of initiation zone organisation
- PMID: 35801867
- PMCID: PMC9303276
- DOI: 10.1093/nar/gkac555
Determination of human DNA replication origin position and efficiency reveals principles of initiation zone organisation
Abstract
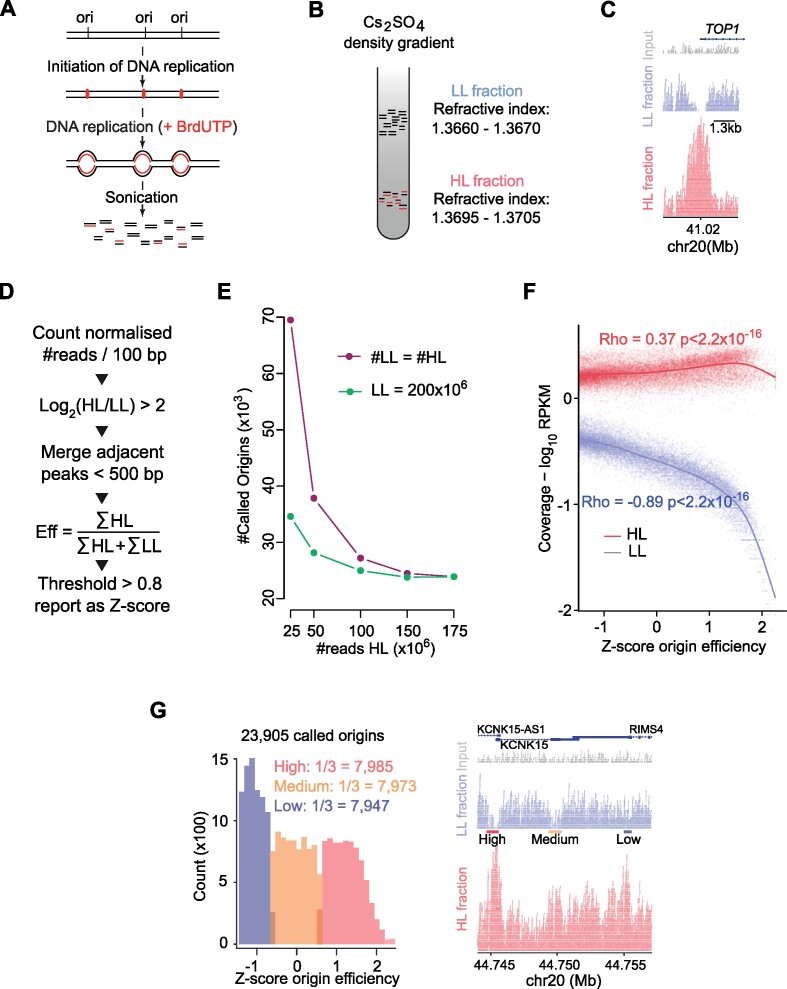
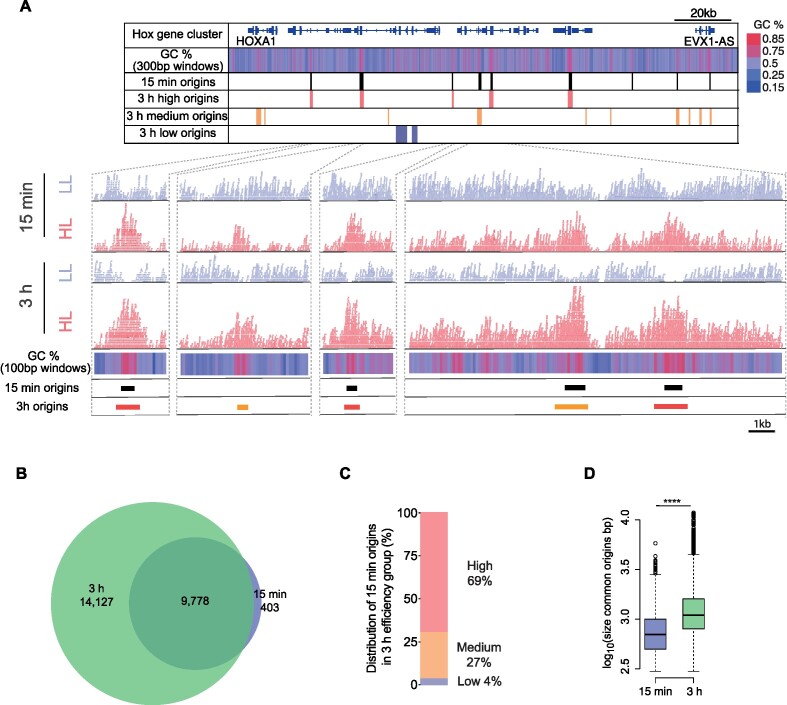
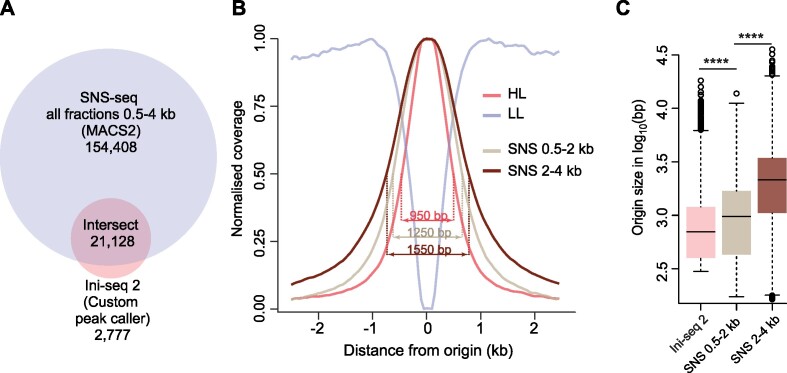
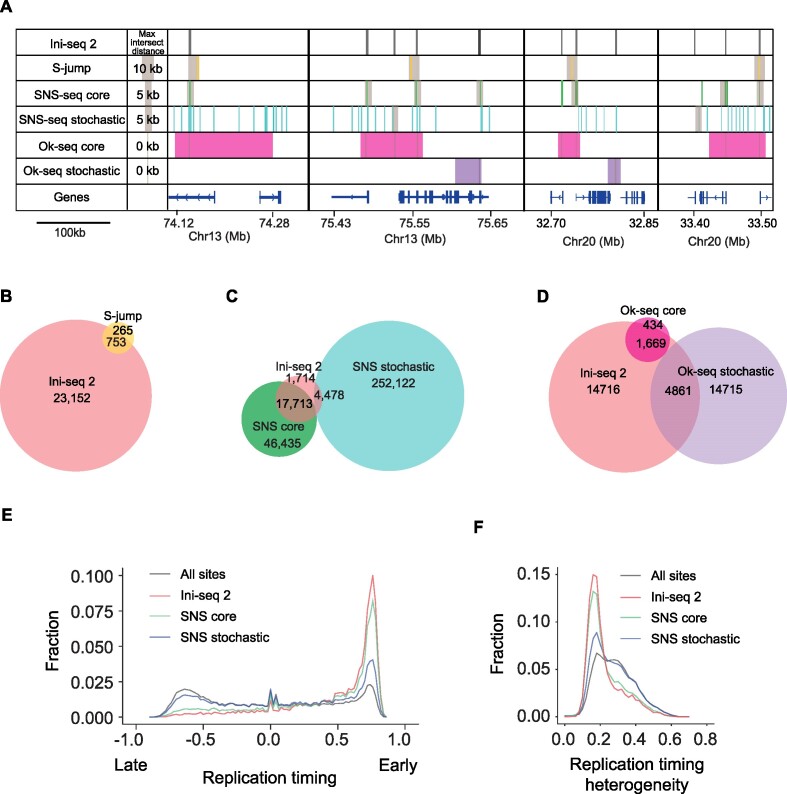
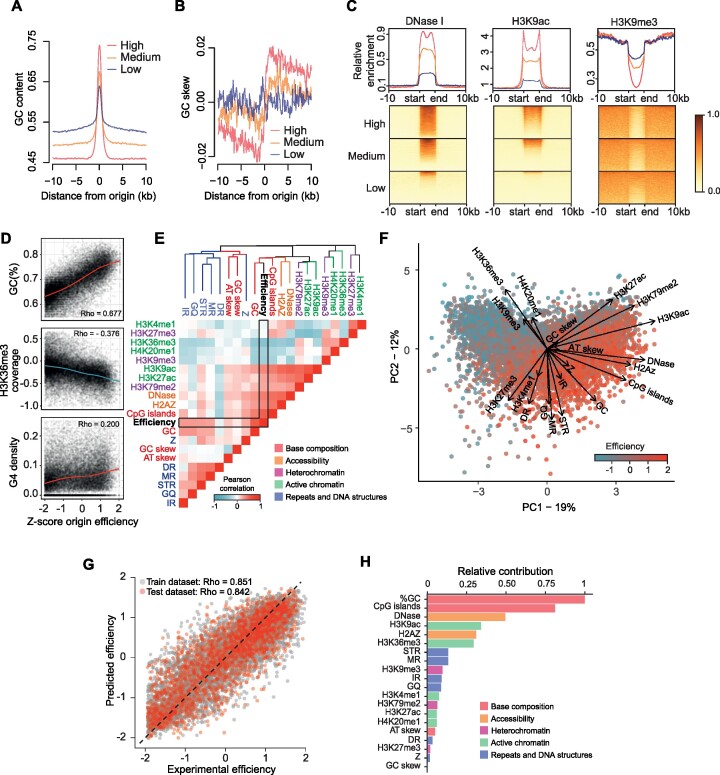
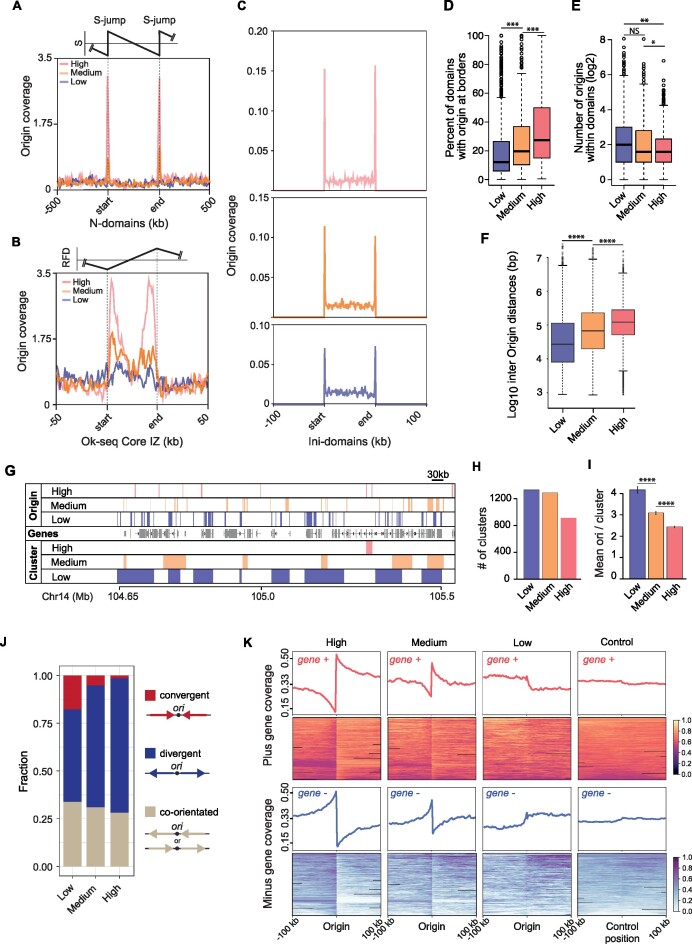
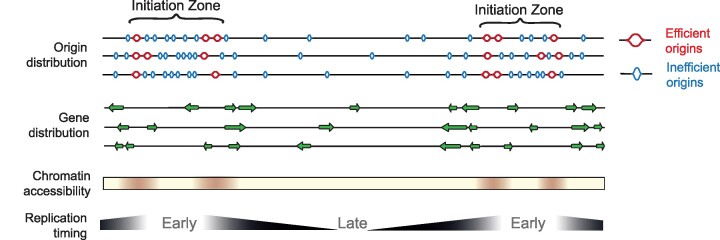
Replication of the human genome initiates within broad zones of ∼150 kb. The extent to which firing of individual DNA replication origins within initiation zones is spatially stochastic or localised at defined sites remains a matter of debate. A thorough characterisation of the dynamic activation of origins within initiation zones is hampered by the lack of a high-resolution map of both their position and efficiency. To address this shortcoming, we describe a modification of initiation site sequencing (ini-seq), based on density substitution. Newly replicated DNA is rendered 'heavy-light' (HL) by incorporation of BrdUTP while unreplicated DNA remains 'light-light' (LL). Replicated HL-DNA is separated from unreplicated LL-DNA by equilibrium density gradient centrifugation, then both fractions are subjected to massive parallel sequencing. This allows precise mapping of 23,905 replication origins simultaneously with an assignment of a replication initiation efficiency score to each. We show that origin firing within early initiation zones is not randomly distributed. Rather, origins are arranged hierarchically with a set of very highly efficient origins marking zone boundaries. We propose that these origins explain much of the early firing activity arising within initiation zones, helping to unify the concept of replication initiation zones with the identification of discrete replication origin sites.
© The Author(s) 2022. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
Genome-wide identification and characterisation of human DNA replication origins by initiation site sequencing (ini-seq).Nucleic Acids Res. 2016 Dec 1;44(21):10230-10247. doi: 10.1093/nar/gkw760. Epub 2016 Sep 1. Nucleic Acids Res. 2016. PMID: 27587586 Free PMC article.
-
DNA replication origins fire stochastically in fission yeast.Mol Biol Cell. 2006 Jan;17(1):308-16. doi: 10.1091/mbc.e05-07-0657. Epub 2005 Oct 26. Mol Biol Cell. 2006. PMID: 16251353 Free PMC article.
-
Identification of primary initiation sites for DNA replication in the hamster dihydrofolate reductase gene initiation zone.Mol Cell Biol. 1998 Jun;18(6):3266-77. doi: 10.1128/MCB.18.6.3266. Mol Cell Biol. 1998. PMID: 9584167 Free PMC article.
-
The double life of mammalian DNA replication origins.Genes Dev. 2025 Mar 3;39(5-6):304-324. doi: 10.1101/gad.352227.124. Genes Dev. 2025. PMID: 39904559 Free PMC article. Review.
-
Replication initiation sites and zones in the mammalian genome: Where are they located and how are they defined?DNA Repair (Amst). 2024 Sep;141:103713. doi: 10.1016/j.dnarep.2024.103713. Epub 2024 Jun 22. DNA Repair (Amst). 2024. PMID: 38959715 Review.
Cited by
-
DNA hypomethylation activates Cdk4/6 and Atr to induce DNA replication and cell cycle arrest to constrain liver outgrowth in zebrafish.Nucleic Acids Res. 2024 Apr 12;52(6):3069-3087. doi: 10.1093/nar/gkae031. Nucleic Acids Res. 2024. PMID: 38321933 Free PMC article.
-
Genome-Wide Mapping of Autonomously Replicating Sequences in the Marine Diatom Phaeodactylum tricornutum.Mar Biotechnol (NY). 2024 Nov 28;27(1):14. doi: 10.1007/s10126-024-10390-0. Mar Biotechnol (NY). 2024. PMID: 39604577
-
Creation and resolution of non-B-DNA structural impediments during replication.Crit Rev Biochem Mol Biol. 2022 Aug;57(4):412-442. doi: 10.1080/10409238.2022.2121803. Epub 2022 Sep 28. Crit Rev Biochem Mol Biol. 2022. PMID: 36170051 Free PMC article.
-
Loss of G1-phase CDK-inhibition biases instability between genomic regions by unevenly reducing activity among replication origins.iScience. 2025 May 28;28(6):112757. doi: 10.1016/j.isci.2025.112757. eCollection 2025 Jun 20. iScience. 2025. PMID: 40546943 Free PMC article.
-
Participants in Transcription-Replication Conflict and Their Role in Formation and Resolution of R-Loops.Int J Mol Sci. 2025 Jul 19;26(14):6951. doi: 10.3390/ijms26146951. Int J Mol Sci. 2025. PMID: 40725198 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
