

Dissecting the treatment-naive ecosystem of human melanoma brain metastasis
- PMID: 35803246
- PMCID: PMC9677434
- DOI: 10.1016/j.cell.2022.06.007
Dissecting the treatment-naive ecosystem of human melanoma brain metastasis
Abstract
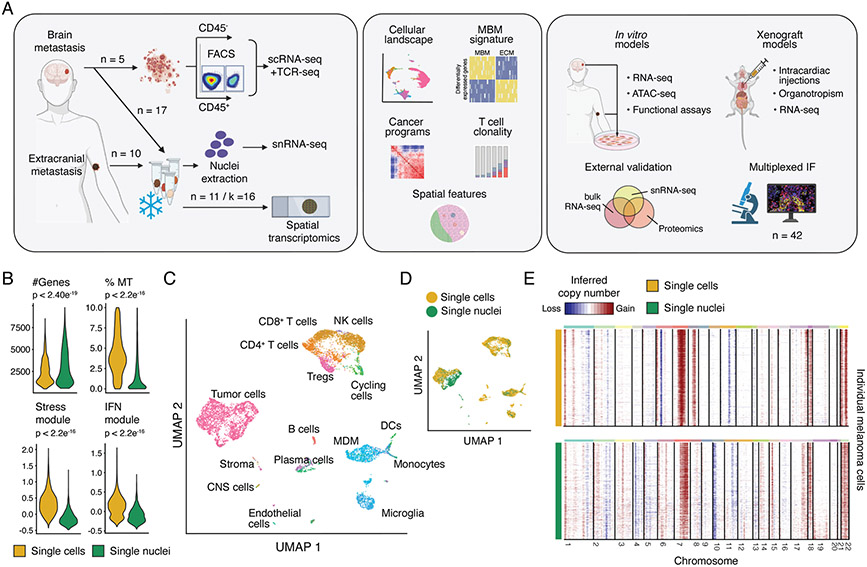
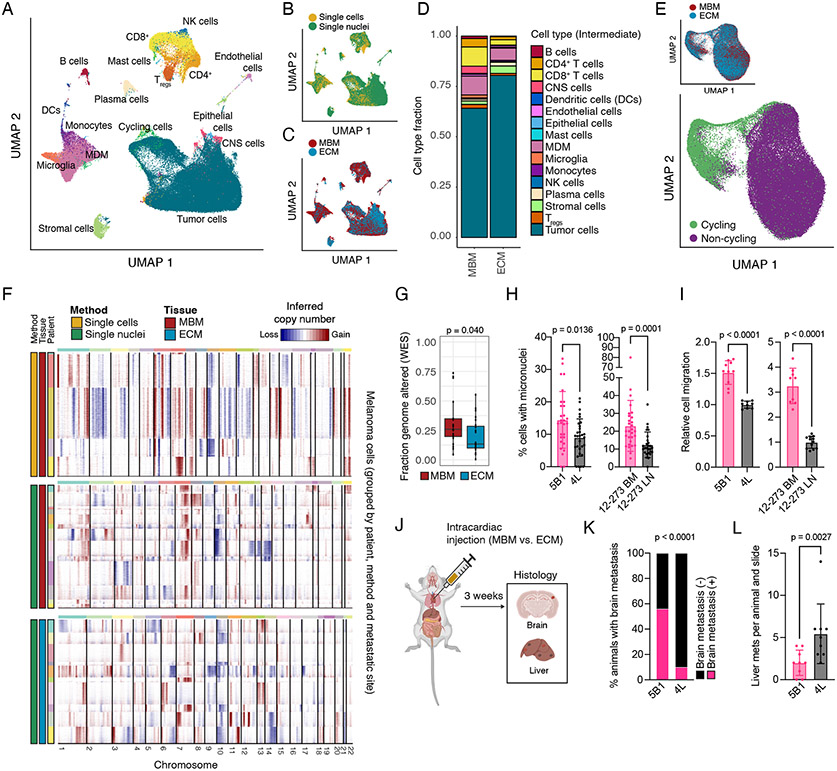
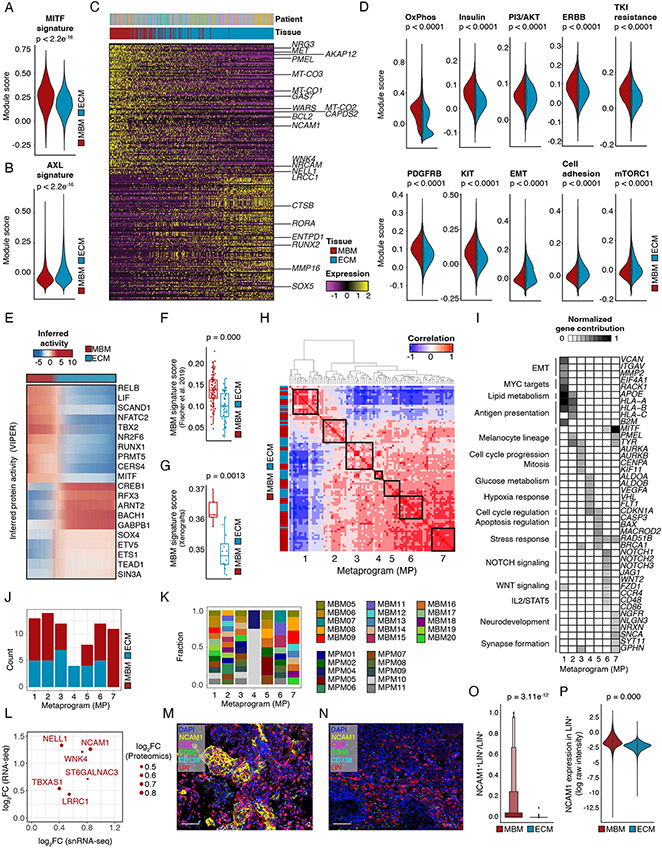
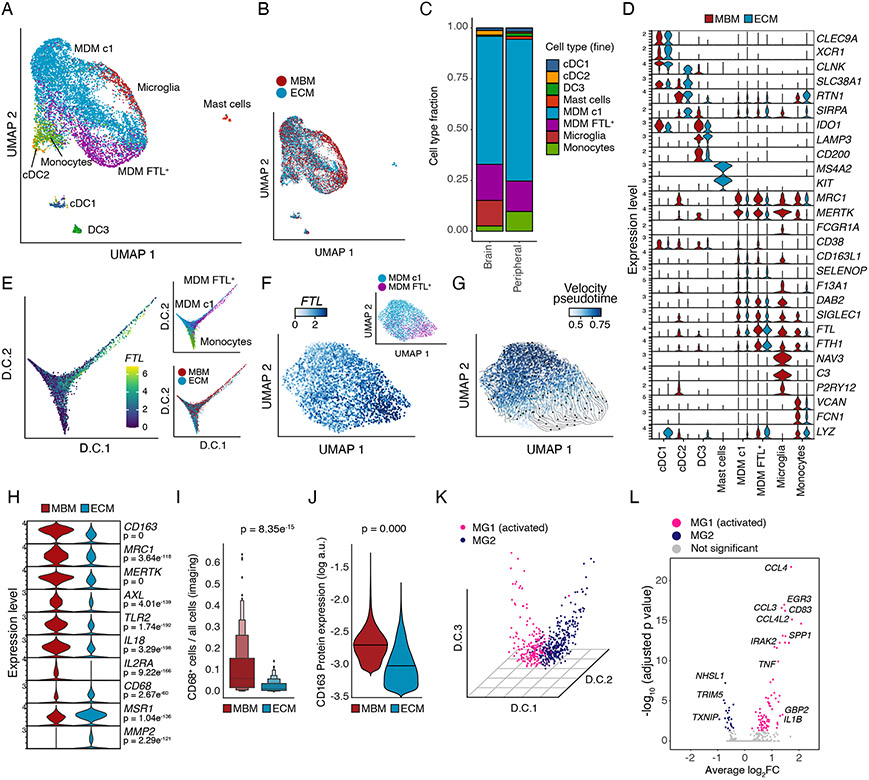
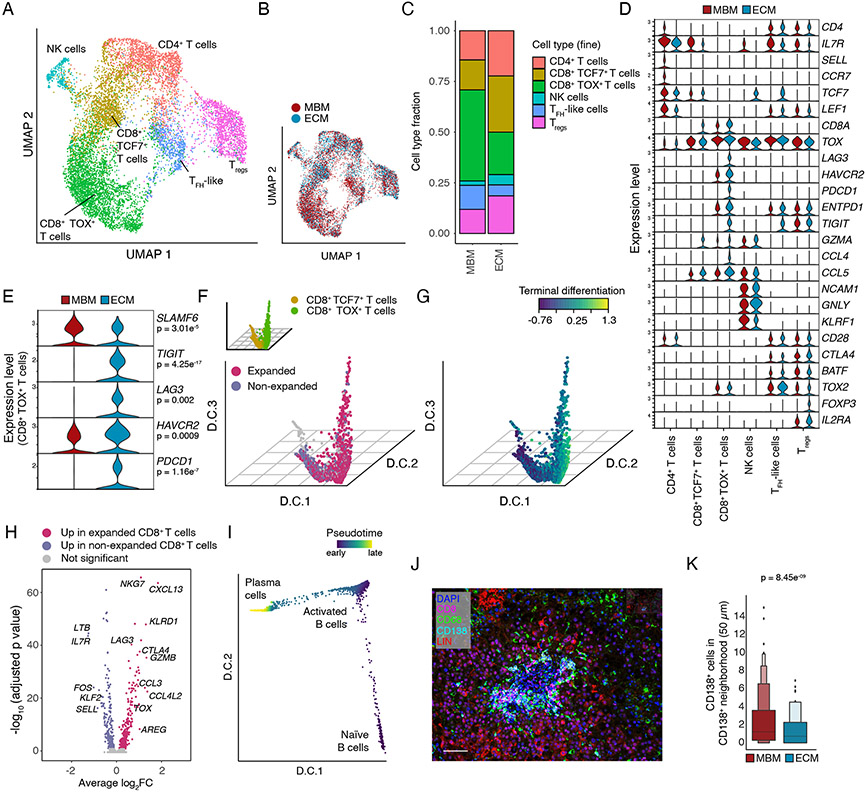
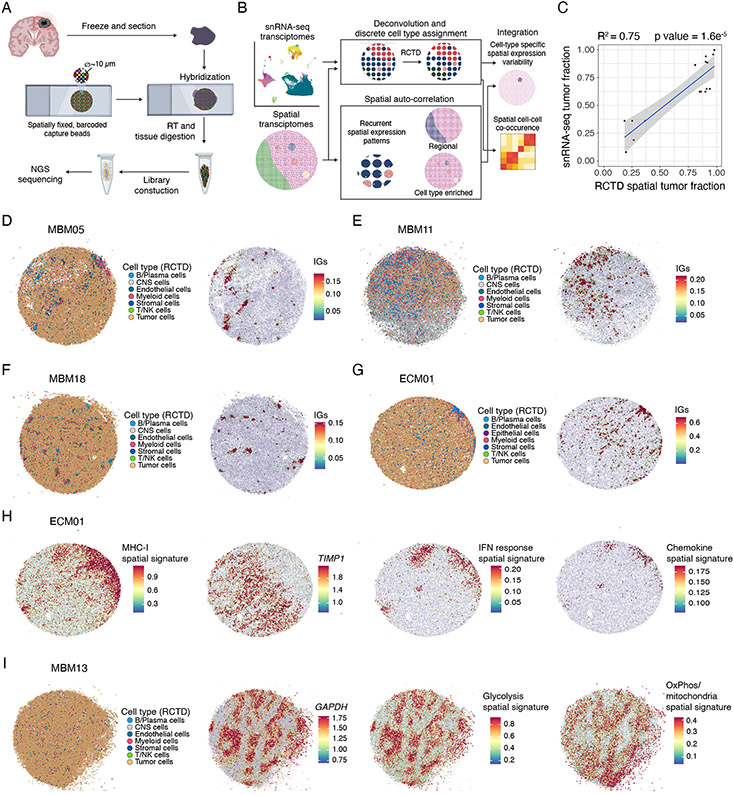
Melanoma brain metastasis (MBM) frequently occurs in patients with advanced melanoma; yet, our understanding of the underlying salient biology is rudimentary. Here, we performed single-cell/nucleus RNA-seq in 22 treatment-naive MBMs and 10 extracranial melanoma metastases (ECMs) and matched spatial single-cell transcriptomics and T cell receptor (TCR)-seq. Cancer cells from MBM were more chromosomally unstable, adopted a neuronal-like cell state, and enriched for spatially variably expressed metabolic pathways. Key observations were validated in independent patient cohorts, patient-derived MBM/ECM xenograft models, RNA/ATAC-seq, proteomics, and multiplexed imaging. Integrated spatial analyses revealed distinct geography of putative cancer immune evasion and evidence for more abundant intra-tumoral B to plasma cell differentiation in lymphoid aggregates in MBM. MBM harbored larger fractions of monocyte-derived macrophages and dysfunctional TOX+CD8+ T cells with distinct expression of immune checkpoints. This work provides comprehensive insights into MBM biology and serves as a foundational resource for further discovery and therapeutic exploration.
Keywords: brain metastasis; chromosomal instability; melanoma; neuronal-like cell state; single-cell genomics; spatial transcriptomics; tumor-microenvironment.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests B.I. has received honoraria from consulting with Merck, Janssen Pharmaceuticals, Astra Zeneca, and Volastra Therapeutics. M.A.D. has been a consultant to Roche/Genentech, Array, Pfizer (New York, NY, United States of America), Novartis, BMS, GSK, Sanofi-Aventis (Bridgewater, NJ, United States of America), Vaccinex, Apexigen, EISAI, and ABM Therapeutics and he has been the PI of research grants to MD Anderson by Roche/Genentech (South San Francisco, CA, United States of America), GSK, Sanofi-Aventis, Merck, Myriad, and Oncothyreon. A.R. has received honoraria from consulting with CStone, Merck, and Vedanta, is or has been a member of the scientific advisory board and holds stock in Advaxis, Appia, Apricity, Arcus, Compugen, CytomX, Highlight, ImaginAb, ImmPact, ImmuneSensor, Inspirna, Isoplexis, Kite-Gilead, Lutris, MapKure, Merus, PACT, Pluto, RAPT, Synthekine, and Tango, has received research funding from Agilent (Santa Clara, CA, United States of America) and from Bristol-Myers Squibb through Stand Up to Cancer (SU2C), and patent royalties from Arsenal Bio. T.E. has acted as a consultant for Almiral Hermal, Bristol-Myers Squibb, MSD, Novartis, Pierre Fabre, and Sanofi. E.Z.M. is a consultant for Curio Bioscience. The other authors do not declare competing interests.
Figures
References
-
- Alberti G (2021). GmAMisc: “Gianmarco Alberti” Miscellaneous. R package version 1.2.0.
Publication types
MeSH terms
Grants and funding
- P30 CA016087/CA/NCI NIH HHS/United States
- T32 GM136573/GM/NIGMS NIH HHS/United States
- T32 GM007367/GM/NIGMS NIH HHS/United States
- K08 CA222663/CA/NCI NIH HHS/United States
- DP5 OD026395/OD/NIH HHS/United States
- P50 CA225450/CA/NCI NIH HHS/United States
- R35 CA197633/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- P30 CA051008/CA/NCI NIH HHS/United States
- U54 CA263001/CA/NCI NIH HHS/United States
- T32 GM145440/GM/NIGMS NIH HHS/United States
- R01 CA256188/CA/NCI NIH HHS/United States
- U54 CA224070/CA/NCI NIH HHS/United States
- T32 AI148099/AI/NIAID NIH HHS/United States
- R37 CA258829/CA/NCI NIH HHS/United States
- U54 CA225088/CA/NCI NIH HHS/United States
- R21 CA263381/CA/NCI NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- P50 CA221703/CA/NCI NIH HHS/United States
- P30 CA013696/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
