

Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy
- PMID: 35803260
- PMCID: PMC9508682
- DOI: 10.1016/j.cell.2022.06.018
Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy
Abstract
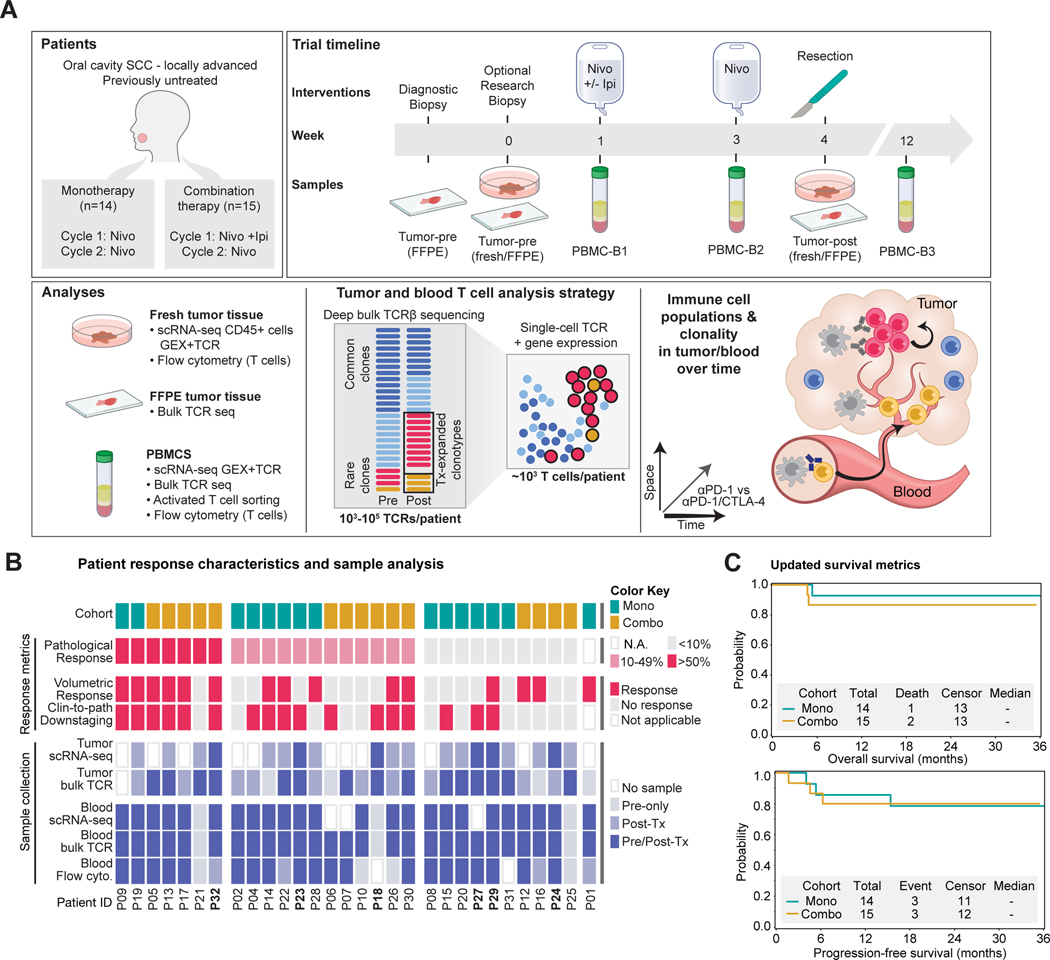
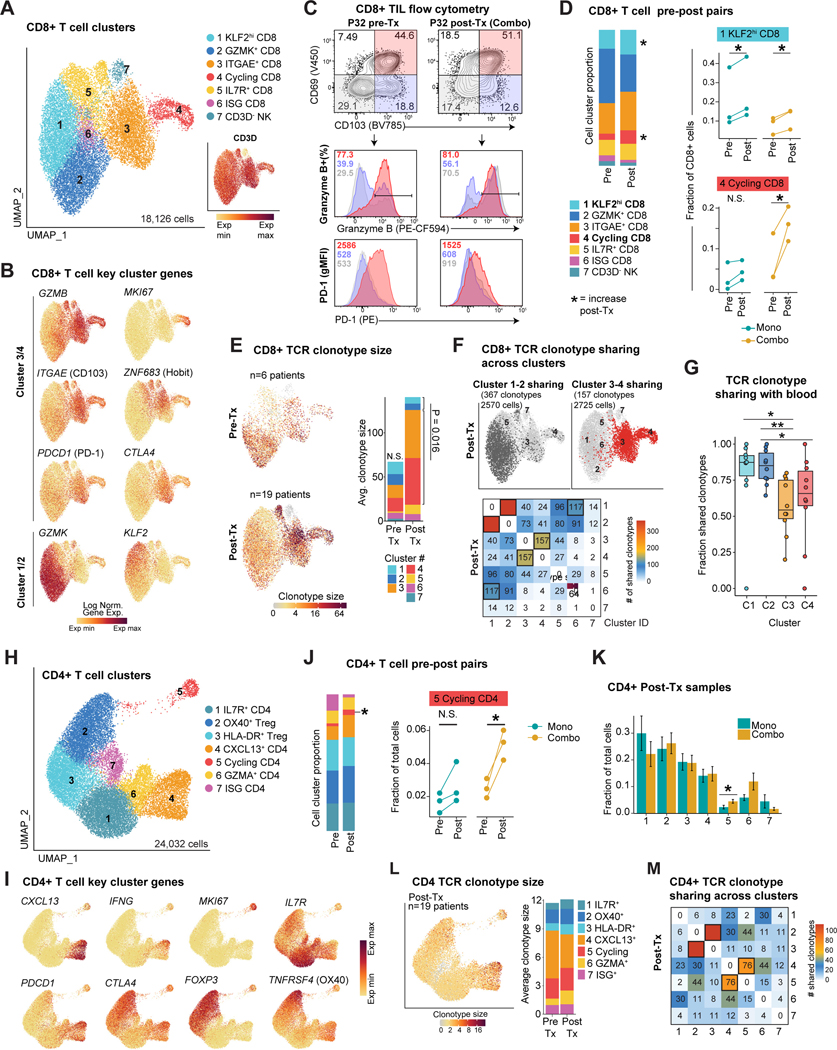
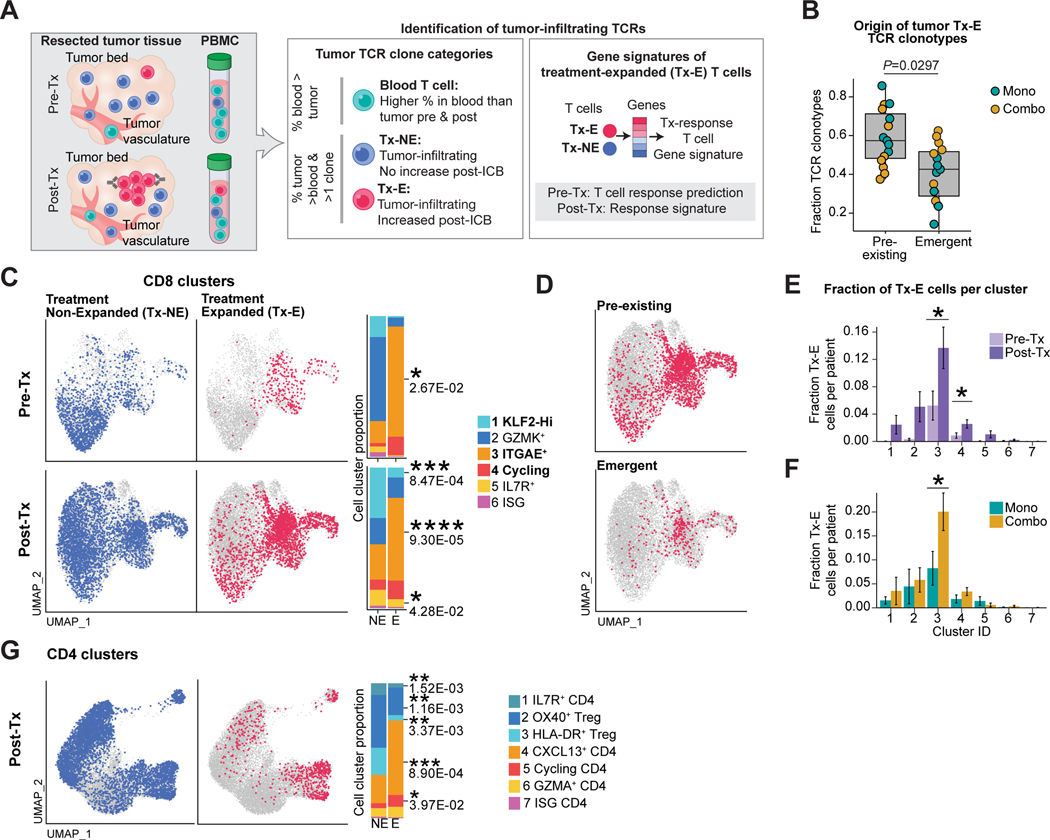
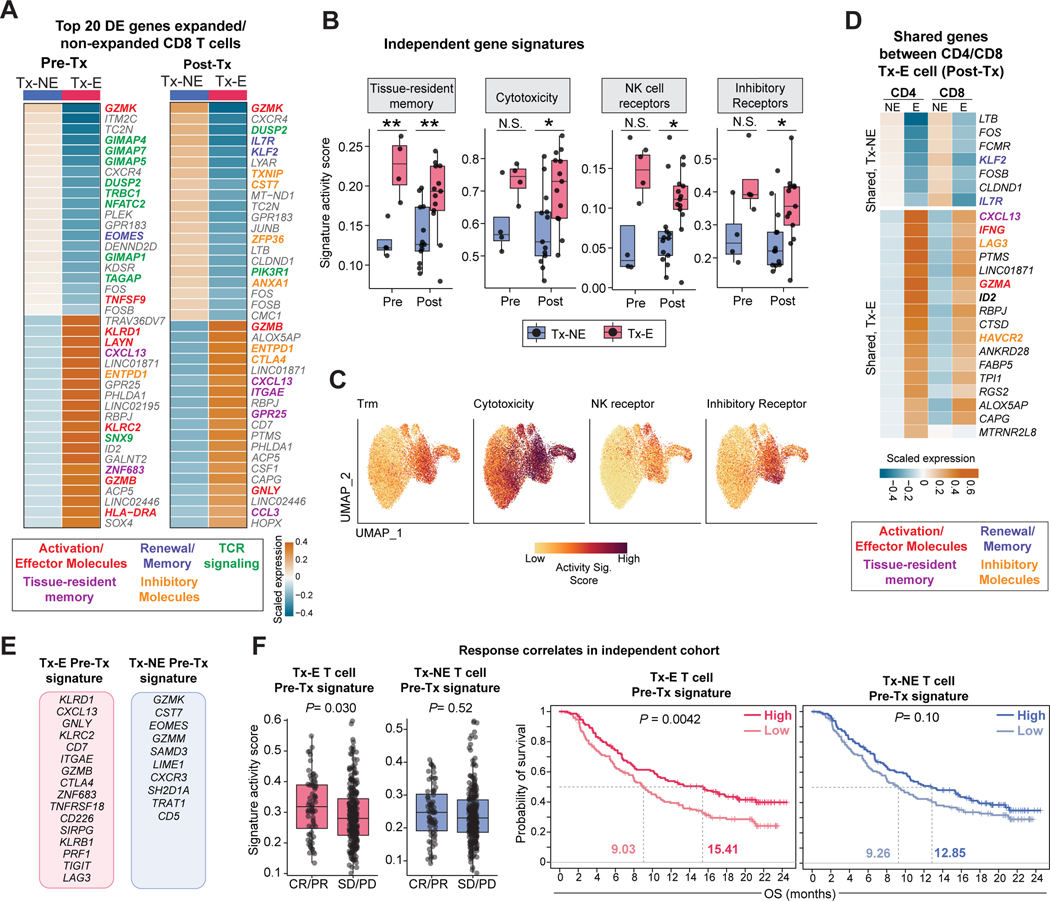
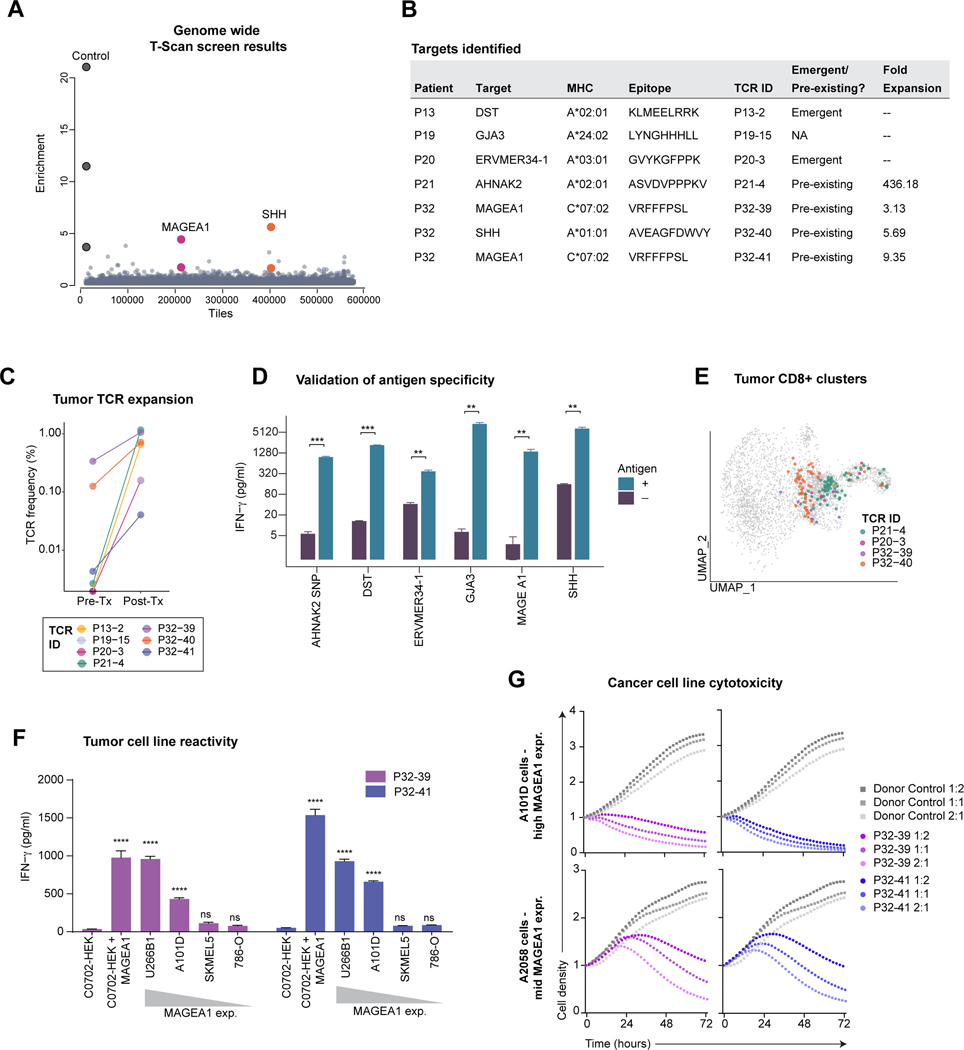
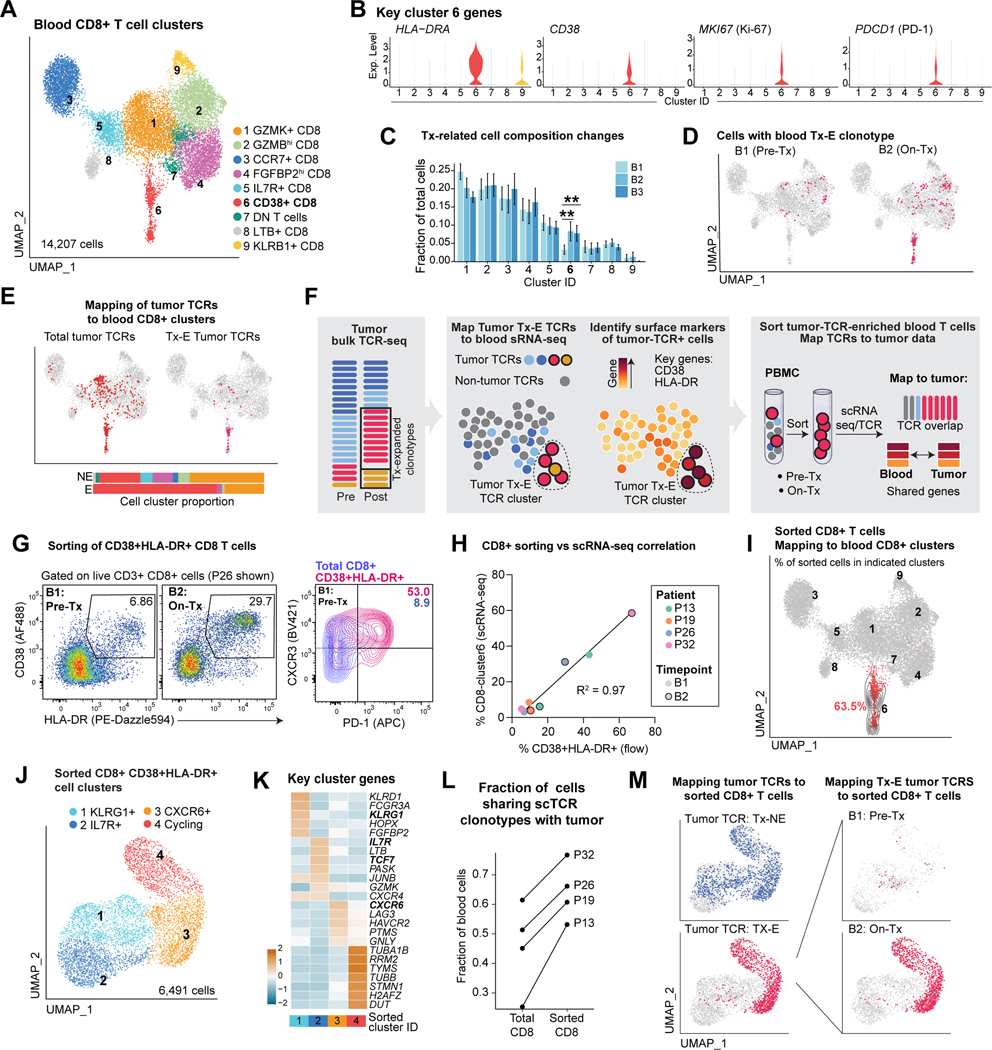
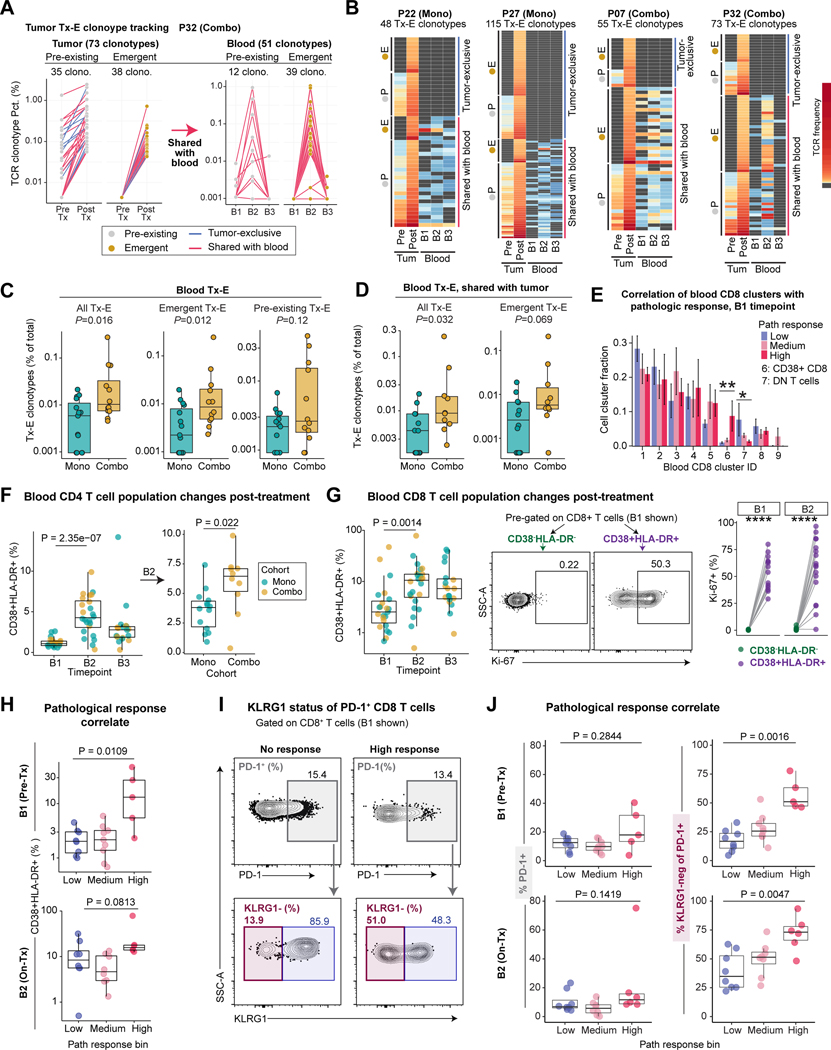
Neoadjuvant immune checkpoint blockade has shown promising clinical activity. Here, we characterized early kinetics in tumor-infiltrating and circulating immune cells in oral cancer patients treated with neoadjuvant anti-PD-1 or anti-PD-1/CTLA-4 in a clinical trial (NCT02919683). Tumor-infiltrating CD8 T cells that clonally expanded during immunotherapy expressed elevated tissue-resident memory and cytotoxicity programs, which were already active prior to therapy, supporting the capacity for rapid response. Systematic target discovery revealed that treatment-expanded tumor T cell clones in responding patients recognized several self-antigens, including the cancer-specific antigen MAGEA1. Treatment also induced a systemic immune response characterized by expansion of activated T cells enriched for tumor-infiltrating T cell clonotypes, including both pre-existing and emergent clonotypes undetectable prior to therapy. The frequency of activated blood CD8 T cells, notably pre-treatment PD-1-positive KLRG1-negative T cells, was strongly associated with intra-tumoral pathological response. These results demonstrate how neoadjuvant checkpoint blockade induces local and systemic tumor immunity.
Keywords: T cells; cancer; immunotherapy; neoadjuvant therapy; single-cell RNA sequencing; tissue-resident memory T cells.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests K.W.W. serves on the SAB of SQZ Biotech, Nextechinvest, Bisou Bioscience Company, and T-Scan Therapeutics and receives sponsored research funding from Novartis. He is a scientific co-founder of Immunitas Therapeutics. J.D.S. reports research support paid to the institution from Merck, BMS, Regeneron, Debiopharm; Consulting/Scientific Advisory Board/Travel fees: Genentech, Immunitas, Debiopharm, BMS, Nanobiotix, Tilos, Castle Biosciences, Astra Zeneca, LEK, Catenion, ACI Clinical, Astellas, Stimit, and Merck KGA; Expert witness fees. Stock options: Immunitas; Equity: Doximity. E.M.V.A. reports Advisory/Consulting: Tango Therapeutics, Genome Medical, Invitae, Enara Bio, Janssen, Manifold Bio, and Monte Rosa; Research support: Novartis, BMS; Equity: Tango Therapeutics, Genome Medical, Syapse, Enara Bio, Manifold Bio, Microsoft, and Monte Rosa; Travel reimbursement: Roche/Genentech; Patents: Institutional patents filed on chromatin mutations and immunotherapy response, and methods for clinical interpretation; intermittent legal consulting on patents for Foaley & Hoag. A.R. is a founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas Therapeutics, and until August 31, 2020, was an SAB member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov, and Thermo Fisher Scientific. From August 1, 2020, A.R. is an employee of Genentech and has equity in Roche.
Figures
Comment in
-
Immune checkpoint blockade provokes resident memory T cells to eliminate head and neck cancer.Cell. 2022 Aug 4;185(16):2848-2849. doi: 10.1016/j.cell.2022.07.006. Cell. 2022. PMID: 35931017
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
