

High levels of NRF2 sensitize temozolomide-resistant glioblastoma cells to ferroptosis via ABCC1/MRP1 upregulation
- PMID: 35803910
- PMCID: PMC9270336
- DOI: 10.1038/s41419-022-05044-9
High levels of NRF2 sensitize temozolomide-resistant glioblastoma cells to ferroptosis via ABCC1/MRP1 upregulation
Abstract
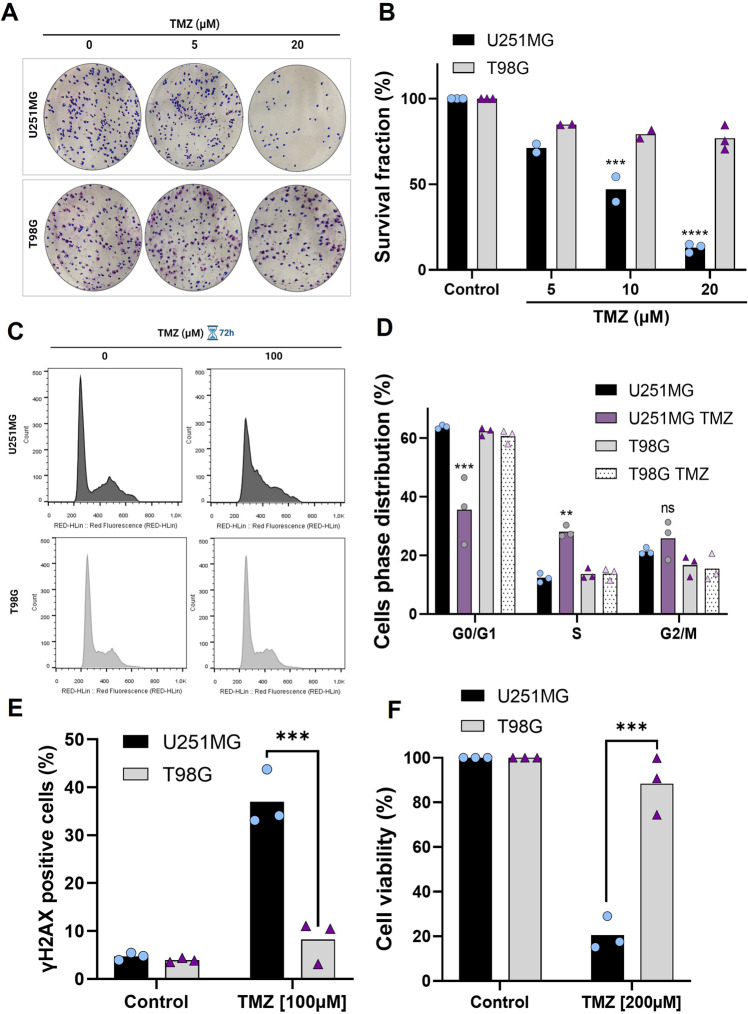
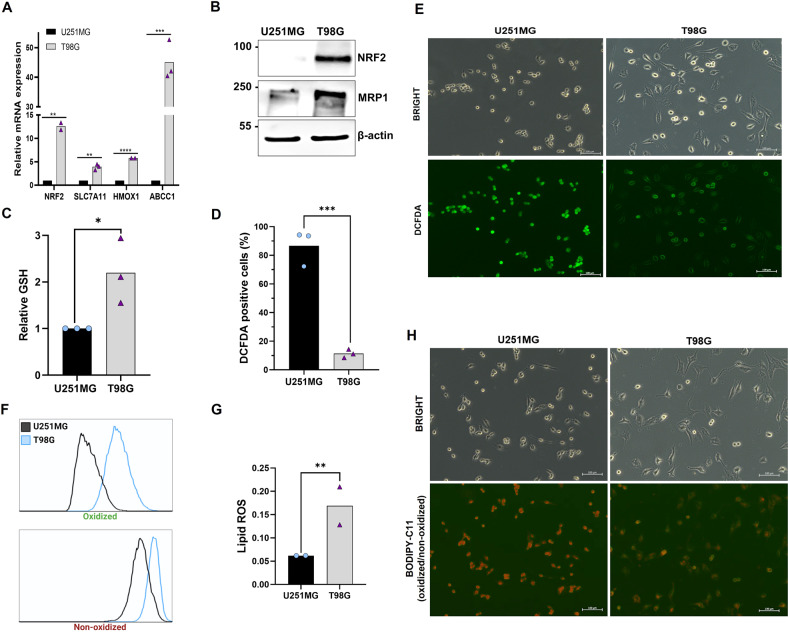
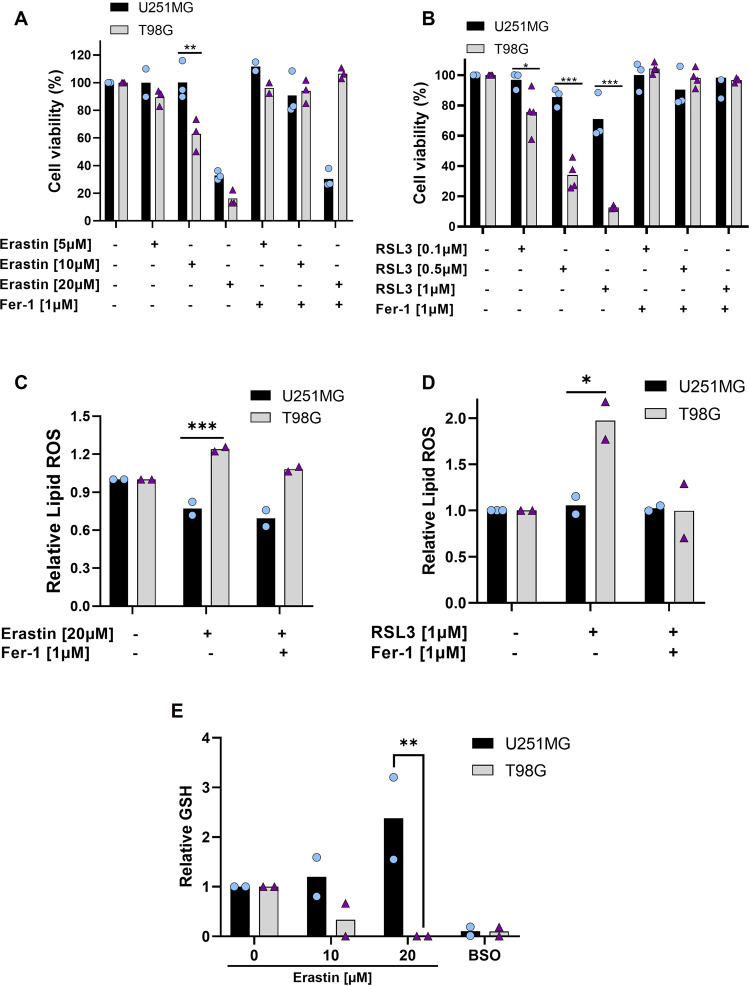
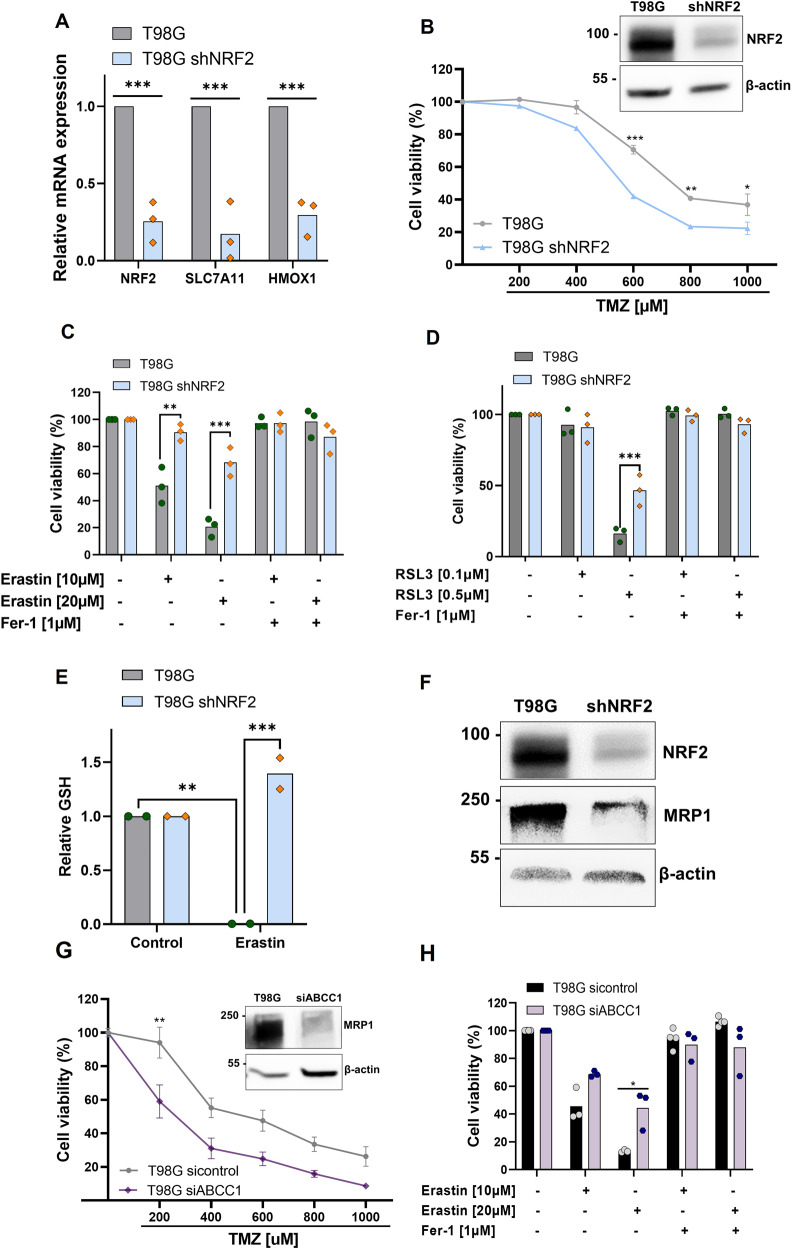
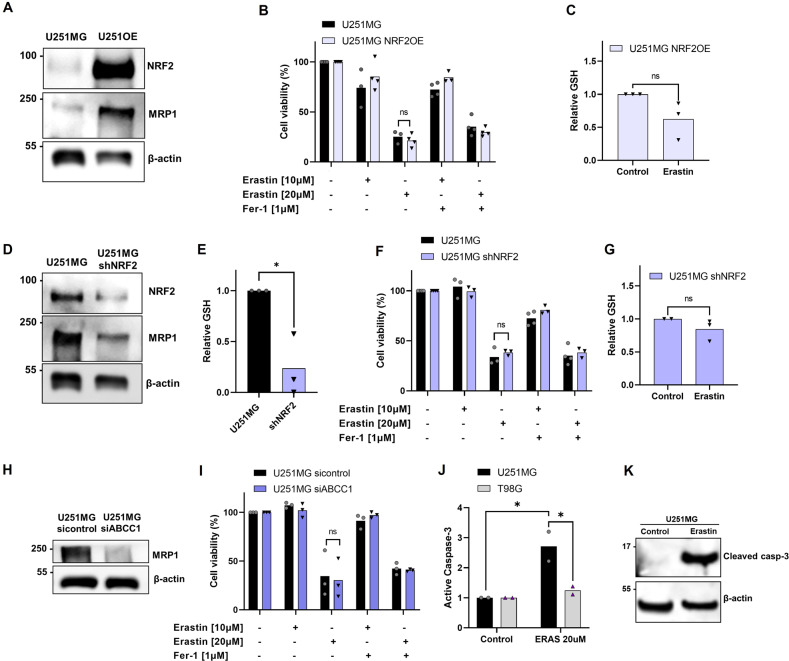
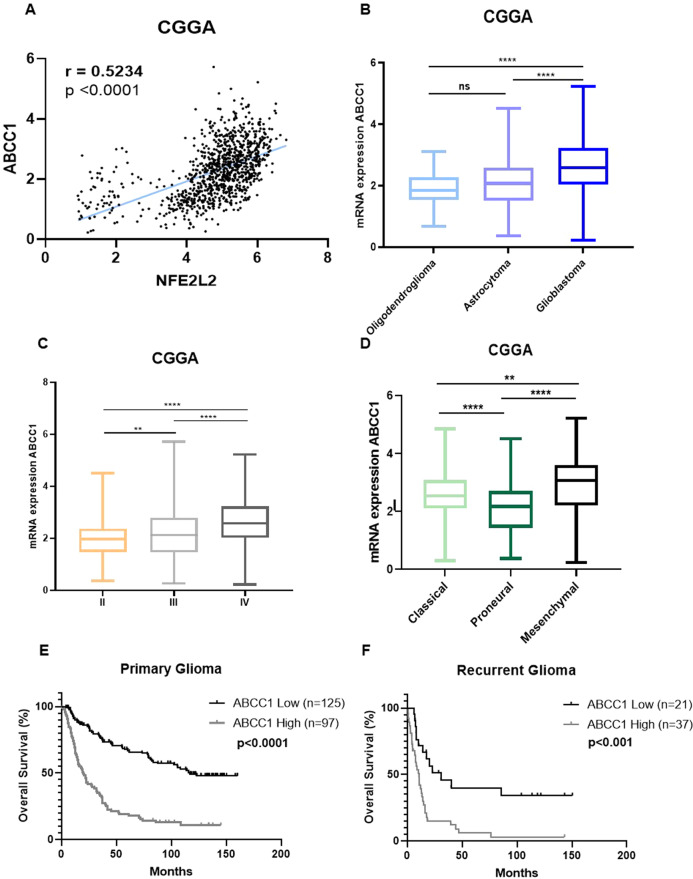
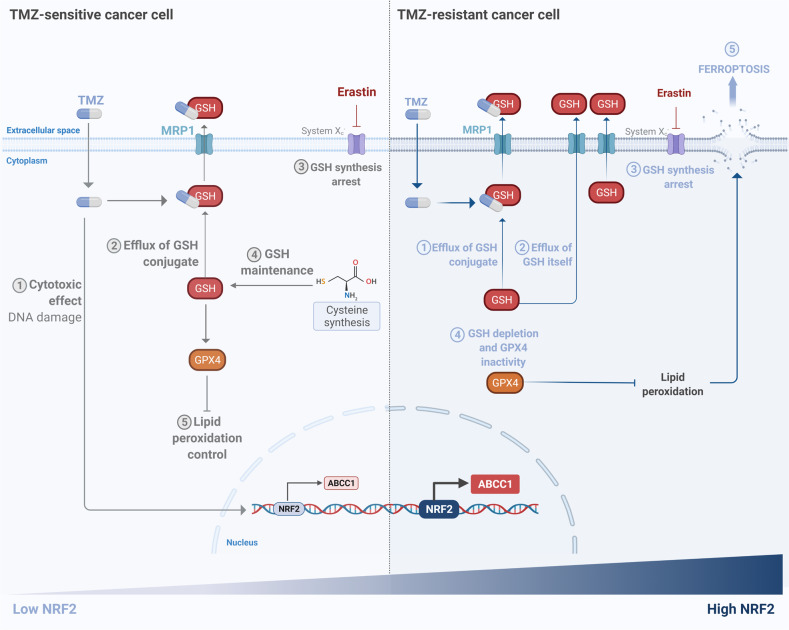
Glioblastoma patients have a poor prognosis mainly due to temozolomide (TMZ) resistance. NRF2 is an important transcript factor involved in chemotherapy resistance due to its protective role in the transcription of genes involved in cellular detoxification and prevention of cell death processes, such as ferroptosis. However, the relation between NRF2 and iron-dependent cell death in glioma is still poorly understood. Therefore, in this study, we analyzed the role of NRF2 in ferroptosis modulation in glioblastoma cells. Two human glioblastoma cell lines (U251MG and T98G) were examined after treatment with TMZ, ferroptosis inducers (Erastin, RSL3), and ferroptosis inhibitor (Ferrostatin-1). Our results demonstrated that T98G was more resistant to chemotherapy compared to U251MG and showed elevated levels of NRF2 expression. Interestingly, T98G revealed higher sensitivity to ferroptosis, and significant GSH depletion upon system xc- blockage. NRF2 silencing in T98G cells (T98G-shNRF2) significantly reduced the viability upon TMZ treatment. On the other hand, T98G-shNRF2 was resistant to ferroptosis and reverted intracellular GSH levels, indicating that NRF2 plays a key role in ferroptosis induction through GSH modulation. Moreover, silencing of ABCC1, a well-known NRF2 target that diminishes GSH levels, has demonstrated a similar collateral sensitivity. T98G-siABCC1 cells were more sensitive to TMZ and resistant to Erastin. Furthermore, we found that NRF2 positively correlates with ABCC1 expression in tumor tissues of glioma patients, which can be associated with tumor aggressiveness, drug resistance, and poor overall survival. Altogether, our data indicate that high levels of NRF2 result in collateral sensitivity on glioblastoma via the expression of its pro-ferroptotic target ABCC1, which contributes to GSH depletion when the system xc- is blocked by Erastin. Thus, ferroptosis induction could be an important therapeutic strategy to reverse drug resistance in gliomas with high NRF2 and ABCC1 expression.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
