

Pro-inflammatory cytokines mediate the epithelial-to-mesenchymal-like transition of pediatric posterior fossa ependymoma
- PMID: 35803925
- PMCID: PMC9270322
- DOI: 10.1038/s41467-022-31683-9
Pro-inflammatory cytokines mediate the epithelial-to-mesenchymal-like transition of pediatric posterior fossa ependymoma
Abstract
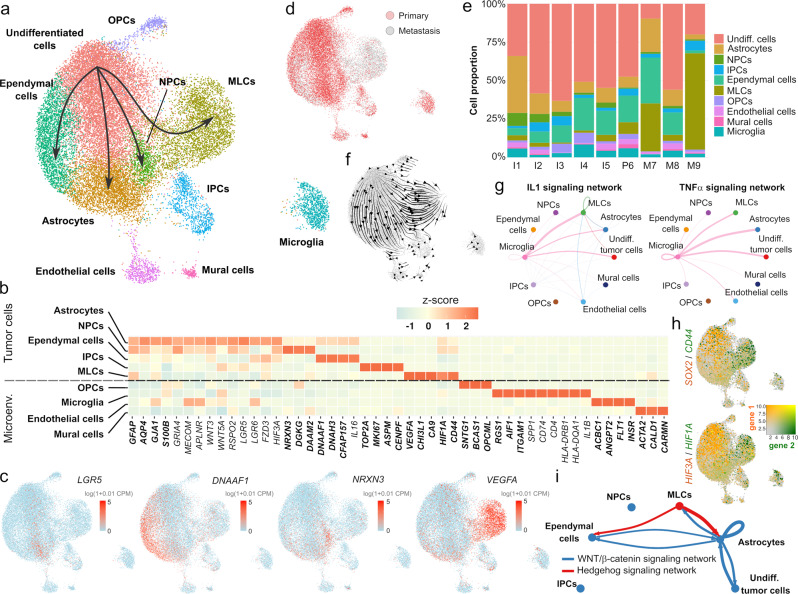
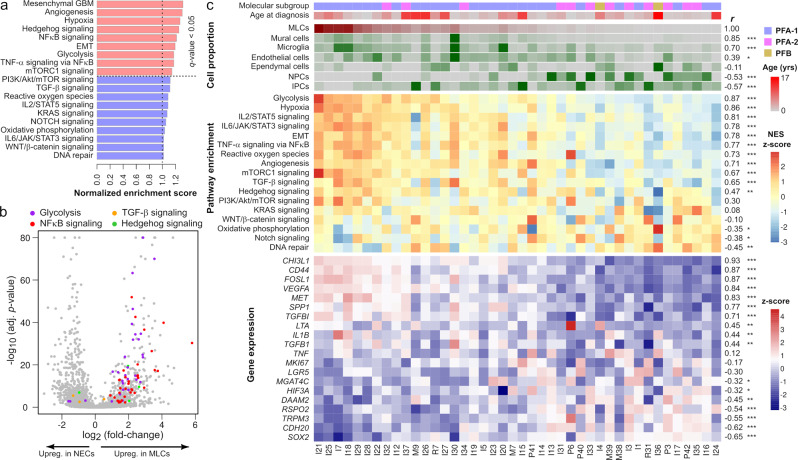
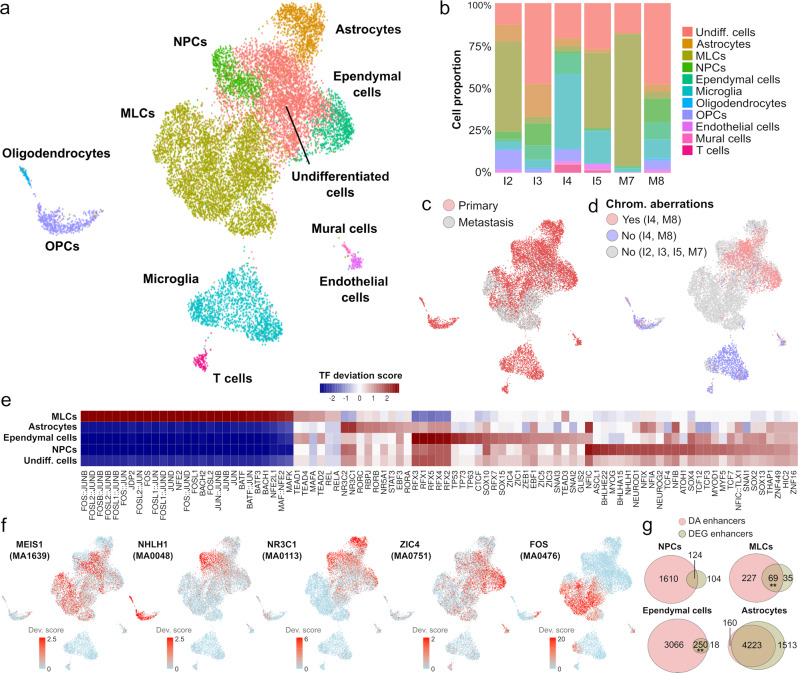
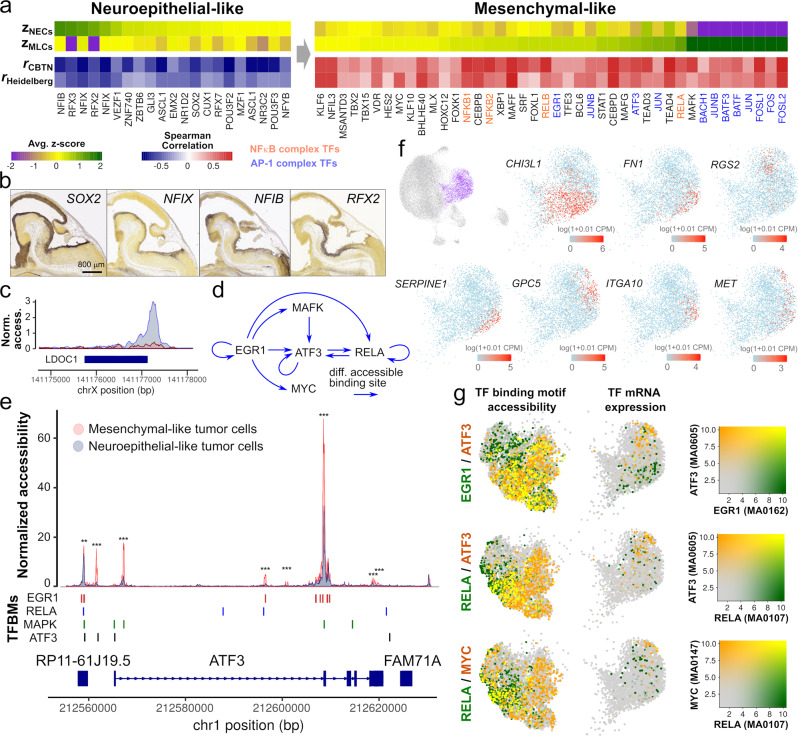
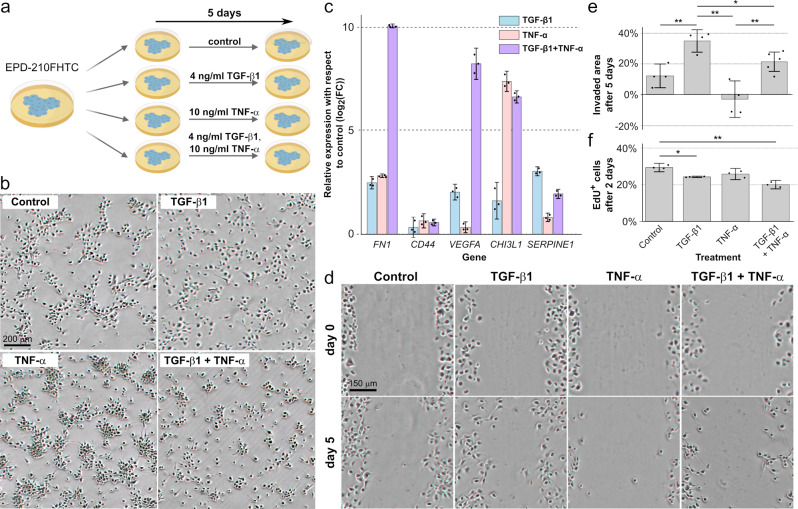
Pediatric ependymoma is a devastating brain cancer marked by its relapsing pattern and lack of effective chemotherapies. This shortage of treatments is due to limited knowledge about ependymoma tumorigenic mechanisms. By means of single-nucleus chromatin accessibility and gene expression profiling of posterior fossa primary tumors and distal metastases, we reveal key transcription factors and enhancers associated with the differentiation of ependymoma tumor cells into tumor-derived cell lineages and their transition into a mesenchymal-like state. We identify NFκB, AP-1, and MYC as mediators of this transition, and show that the gene expression profiles of tumor cells and infiltrating microglia are consistent with abundant pro-inflammatory signaling between these populations. In line with these results, both TGF-β1 and TNF-α induce the expression of mesenchymal genes on a patient-derived cell model, and TGF-β1 leads to an invasive phenotype. Altogether, these data suggest that tumor gliosis induced by inflammatory cytokines and oxidative stress underlies the mesenchymal phenotype of posterior fossa ependymoma.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
