

dia-PASEF data analysis using FragPipe and DIA-NN for deep proteomics of low sample amounts
- PMID: 35803928
- PMCID: PMC9270362
- DOI: 10.1038/s41467-022-31492-0
dia-PASEF data analysis using FragPipe and DIA-NN for deep proteomics of low sample amounts
Abstract
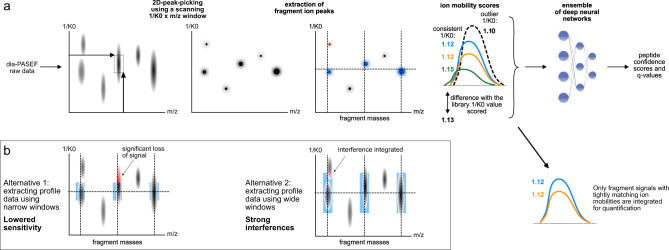
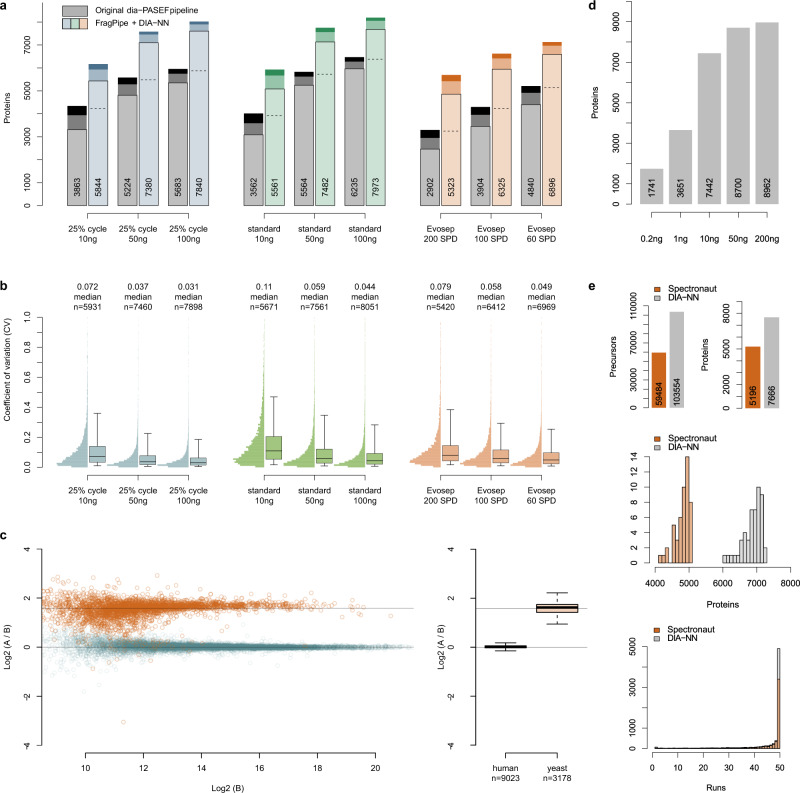
The dia-PASEF technology uses ion mobility separation to reduce signal interferences and increase sensitivity in proteomic experiments. Here we present a two-dimensional peak-picking algorithm and generation of optimized spectral libraries, as well as take advantage of neural network-based processing of dia-PASEF data. Our computational platform boosts proteomic depth by up to 83% compared to previous work, and is specifically beneficial for fast proteomic experiments and those with low sample amounts. It quantifies over 5300 proteins in single injections recorded at 200 samples per day throughput using Evosep One chromatography system on a timsTOF Pro mass spectrometer and almost 9000 proteins in single injections recorded with a 93-min nanoflow gradient on timsTOF Pro 2, from 200 ng of HeLa peptides. A user-friendly implementation is provided through the incorporation of the algorithms in the DIA-NN software and by the FragPipe workflow for spectral library generation.
© 2022. The Author(s).
Conflict of interest statement
J.D. and S.K.-S. are employees of Bruker Daltonics. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
