

The cholesterol uptake regulator PCSK9 promotes and is a therapeutic target in APC/KRAS-mutant colorectal cancer
- PMID: 35803966
- PMCID: PMC9270407
- DOI: 10.1038/s41467-022-31663-z
The cholesterol uptake regulator PCSK9 promotes and is a therapeutic target in APC/KRAS-mutant colorectal cancer
Abstract
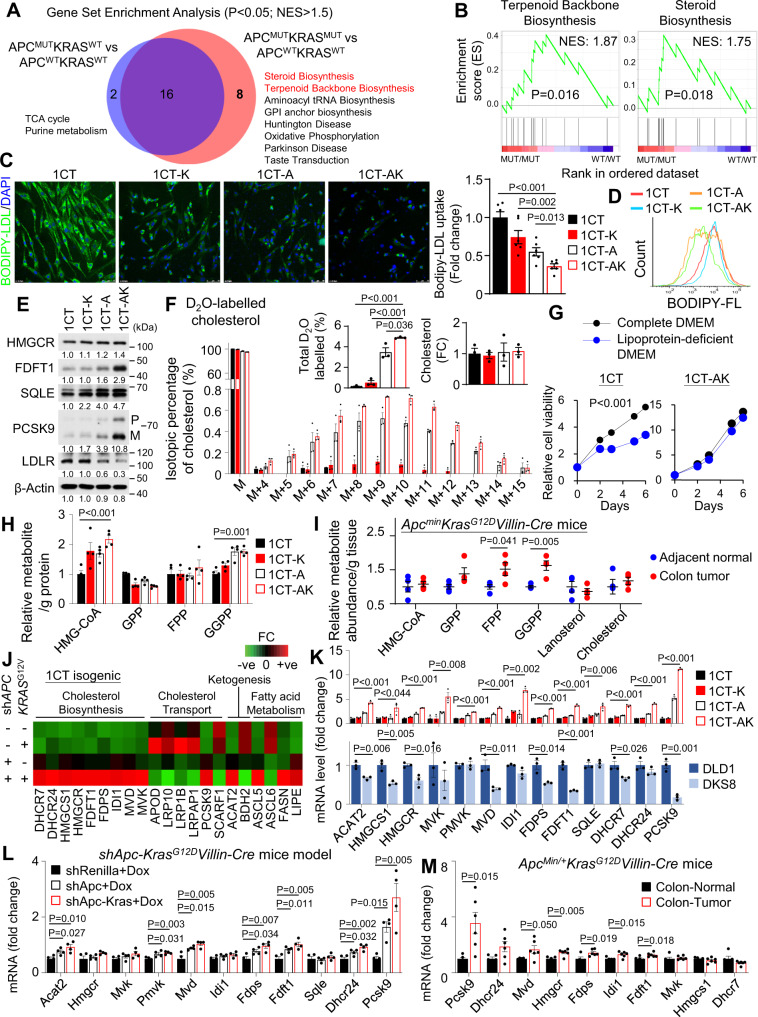
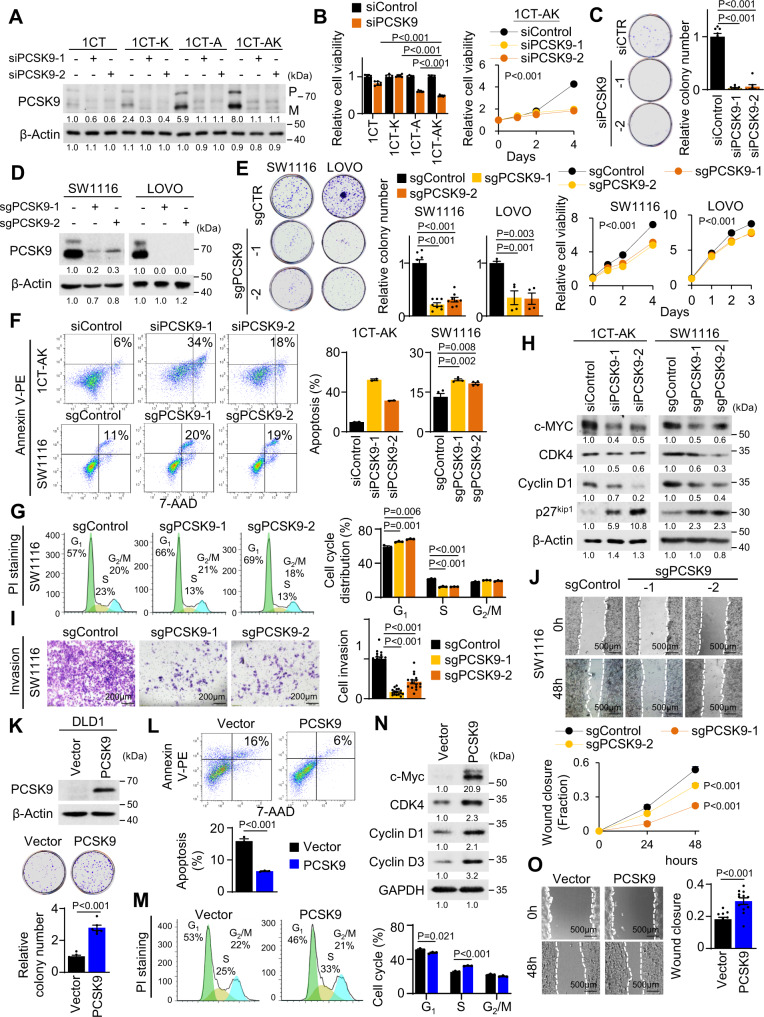
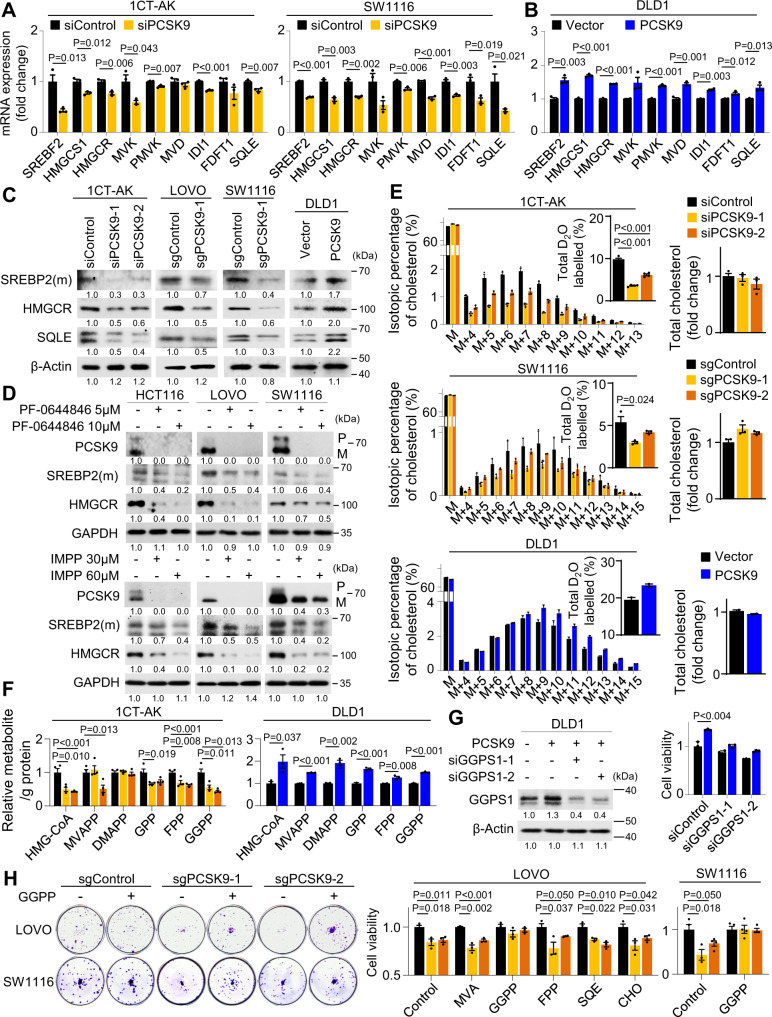
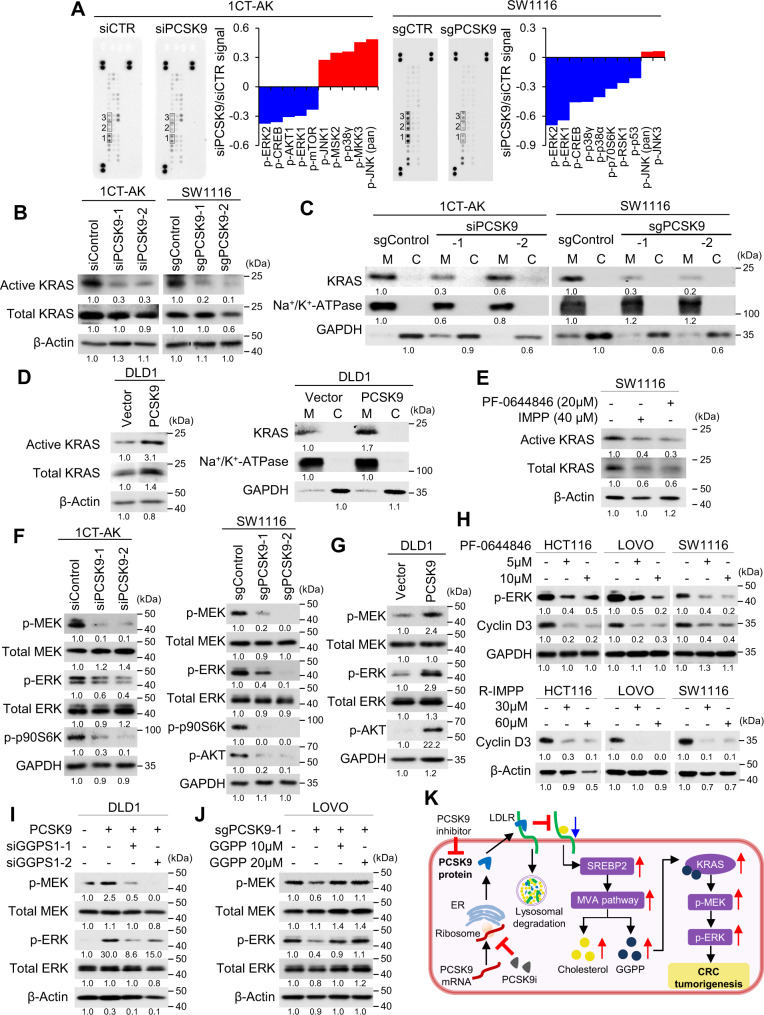
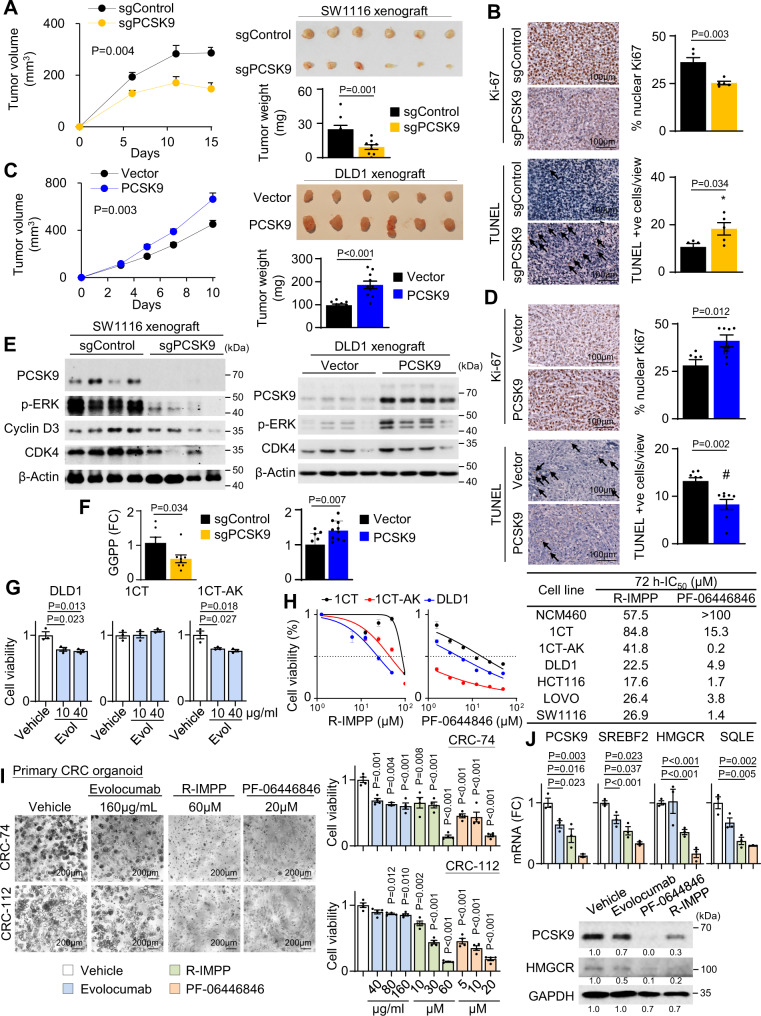
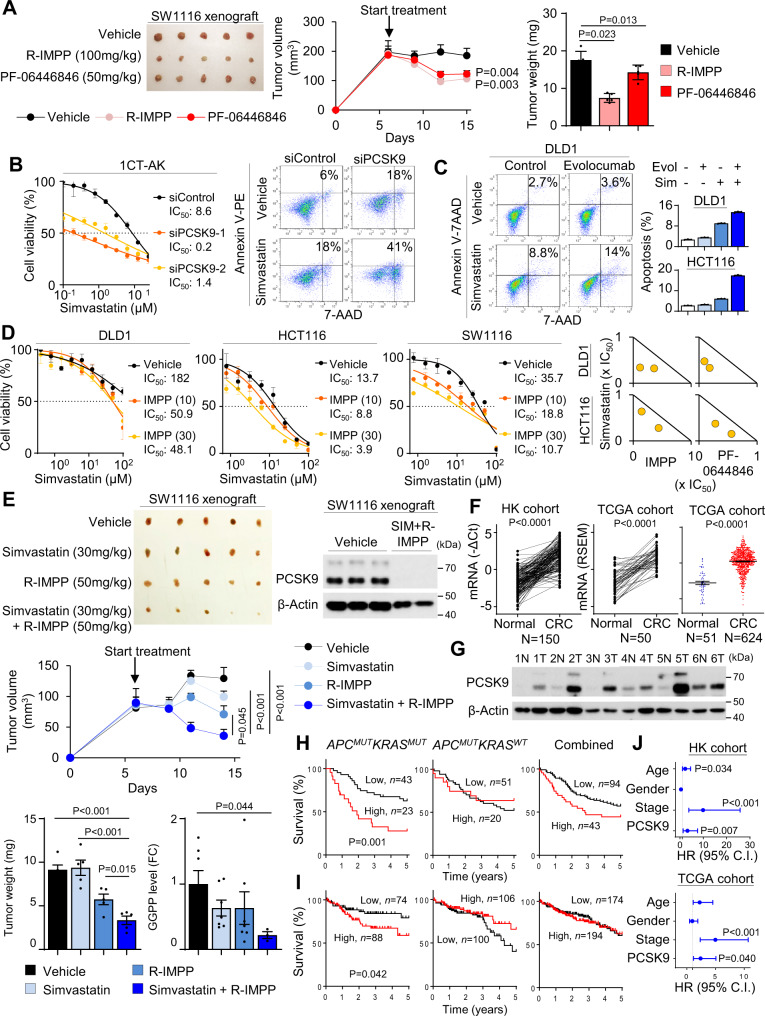
Therapeutic targeting of KRAS-mutant colorectal cancer (CRC) is an unmet need. Here, we show that Proprotein Convertase Subtilisin/Kexin type 9 (PSCK9) promotes APC/KRAS-mutant CRC and is a therapeutic target. Using CRC patient cohorts, isogenic cell lines and transgenic mice, we identify that de novo cholesterol biosynthesis is induced in APC/KRAS mutant CRC, accompanied by increased geranylgeranyl diphosphate (GGPP)─a metabolite necessary for KRAS activation. PCSK9 is the top up-regulated cholesterol-related gene. PCSK9 depletion represses APC/KRAS-mutant CRC cell growth in vitro and in vivo, whereas PCSK9 overexpression induces oncogenesis. Mechanistically, PCSK9 reduces cholesterol uptake but induces cholesterol de novo biosynthesis and GGPP accumulation. GGPP is a pivotal metabolite downstream of PCSK9 by activating KRAS/MEK/ERK signaling. PCSK9 inhibitors suppress growth of APC/KRAS-mutant CRC cells, organoids and xenografts, especially in combination with simvastatin. PCSK9 overexpression predicts poor survival of APC/KRAS-mutant CRC patients. Together, cholesterol homeostasis regulator PCSK9 promotes APC/KRAS-mutant CRC via GGPP-KRAS/MEK/ERK axis and is a therapeutic target.
© 2022. The Author(s).
Conflict of interest statement
The authors declared no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
