

High childhood serum triglyceride concentrations associate with hepatocellular adenoma development in patients with glycogen storage disease type Ia
- PMID: 35811762
- PMCID: PMC9263528
- DOI: 10.1016/j.jhepr.2022.100512
High childhood serum triglyceride concentrations associate with hepatocellular adenoma development in patients with glycogen storage disease type Ia
Abstract
Background & aims: Glycogen storage disease type Ia (GSDIa) is an inborn error of carbohydrate metabolism caused by pathogenic variants in the glucose-6-phosphatase catalytic subunit 1 (G6PC1) gene and is associated with hepatocellular adenoma (HCA) formation. Data on risk factors for HCA occurrence in GSDIa are scarce. We investigated HCA development in relation to sex, G6PC1 genotype, and serum triglyceride concentration (TG).
Methods: An observational study of patients with genetically confirmed GSDIa ≥12 years was performed. Patients were categorised for sex; presence of 2, 1, or 0 predicted severe G6PC1 variant (PSV); and median TG during childhood (<12 years; stratified for above/below 5.65 mmol/L, i.e. 500 mg/dl).
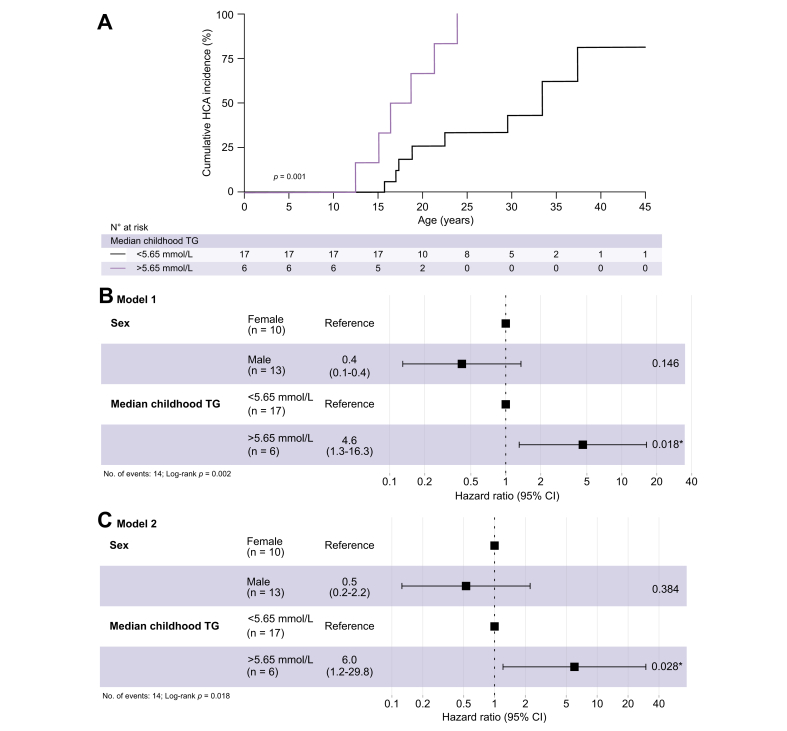
Results: Fifty-three patients (23 females) were included, of which 26 patients developed HCA at a median (IQR) age of 21 (17-25) years. At the age of 25 years, 48% of females and 30% of males had developed HCA (log-rank p = 0.045). Two-thirds of patients with GSDIa carried 2 PSVs, 20% carried 1, and 13% carried none. Neither the number of PSVs nor any specific G6PC1 variants were associated with HCA occurrence. Childhood TG was 3.4 (3.0-4.2) mmol/L in males vs. 5.6 (4.0-7.9) mmol/L in females (p = 0.026). Childhood TG >5.65 mmol/L was associated with HCA development at younger age, compared with patients with childhood TG <5.65 mmol/L (18 vs. 33 years; log-rank p = 0.001). Cox regression analysis including TG, sex, and TG-sex interaction correction revealed childhood TG >5.65 mmol/L as an independent risk factor for HCA development (hazard ratio [HR] 6.0; 95% CI 1.2-29.8; p = 0.028).
Conclusions: In patients with GSDIa, high childhood TG was associated with an increased risk of HCA, and earlier onset of HCA development, independent of sex-associated hypertriglyceridaemia, and G6PC1 genotype.
Lay summary: Glycogen storage disease type Ia (GSDIa) is a rare, inherited metabolic disease that can be complicated by liver tumours (hepatocellular adenomas), which in turn may cause bleeding or progress to liver cancer. Risk factors associated with hepatocellular adenoma formation in patients with GSDIa are largely unknown. In our study, we found that high serum triglyceride concentrations during childhood, but not specific genetic variants, were associated with increased risk of hepatocellular adenoma diagnosis later in life.
Keywords: Benign neoplasm; G6PC1, glucose-6-phosphatase catalytic subunit 1; G6Pase, glucose-6-phosphatase; GSDIa, glycogen storage disease type Ia; Glucose-6-phosphatase triglycerides; Glycogen storage disease type Ia; HCA, hepatocellular adenoma; HR, hazard ratio; Hepatic adenoma; MRI, magnetic resonance imaging; Metabolic disorder; PSV, predicted severe variant; TG, serum triglyceride concentration.
© 2022 The Author(s).
Conflict of interest statement
There are no conflicts of interest to be reported. Please refer to the accompanying ICMJE disclosure forms for further details.
Figures




References
-
- Kishnani P.S., Austin S.L., Abdenur J.E., Arn P., Bali D.S., Boney A., et al. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet Med. 2014;16:e1–e29. - PubMed
-
- Rake J., Visser G., Labrune P., Leonard J., Ullrich K., Smit P. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I) Eur J Pediatr. 2002;161:S20–S34. - PubMed
-
- Rake J.P., Visser G., Labrune P., Leonard J.V., Ullrich K., Smit G.P.A., et al. Guidelines for management of glycogen storage disease type I – European Study on Glycogen Storage Disease Type I (ESGSD I) Eur J Pediatr. 2002;161(Suppl. 1):S112–S119. - PubMed
-
- Bali D.S., El-Gharbawy A., Austin S., Pendyal S., Kishnani P.S. In: Adam M.P., Mirzaa G.M., Pagon R.A., et al., editors. University of Seattle; Seattle (WA): 2021. Glycogen storage disease type I. GeneReviews®.https://www.ncbi.nlm.nih.gov/books/NBK1312/ - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
