

Peptidome Surveillance Across Evolving SARS-CoV-2 Lineages Reveals HLA Binding Conservation in Nucleocapsid Among Variants With Most Potential for T-Cell Epitope Loss in Spike
- PMID: 35812370
- PMCID: PMC9260595
- DOI: 10.3389/fimmu.2022.918928
Peptidome Surveillance Across Evolving SARS-CoV-2 Lineages Reveals HLA Binding Conservation in Nucleocapsid Among Variants With Most Potential for T-Cell Epitope Loss in Spike
Abstract
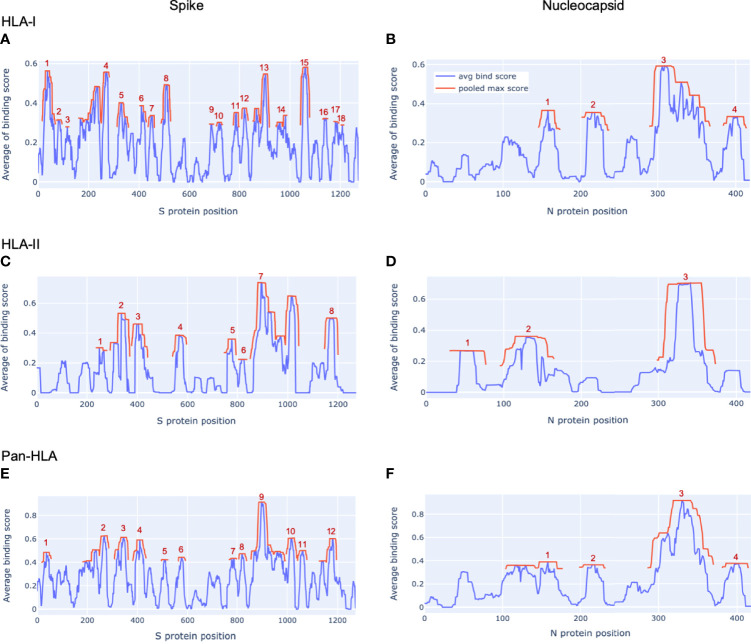
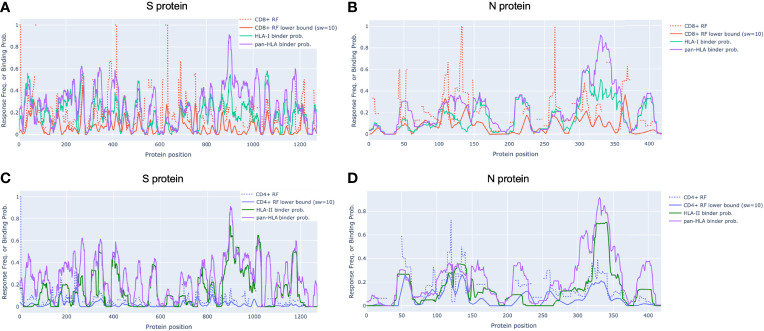
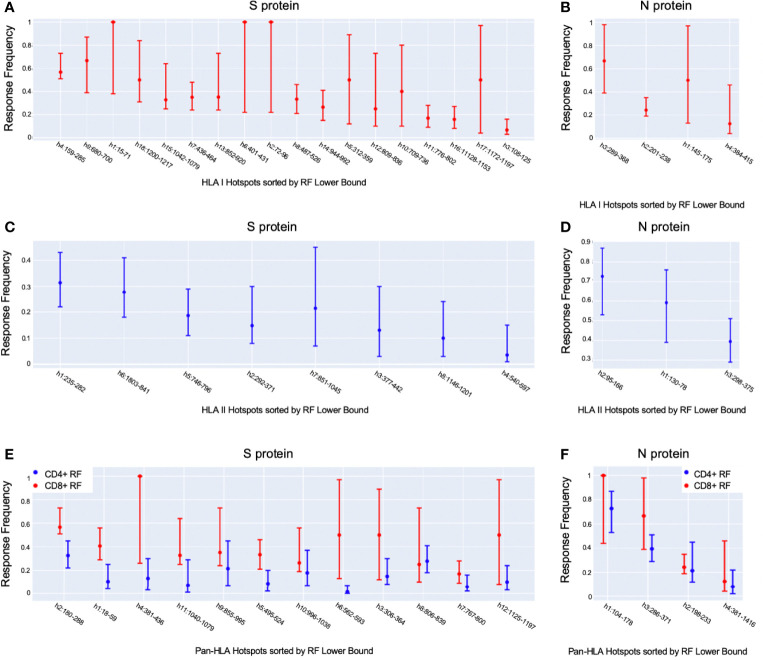
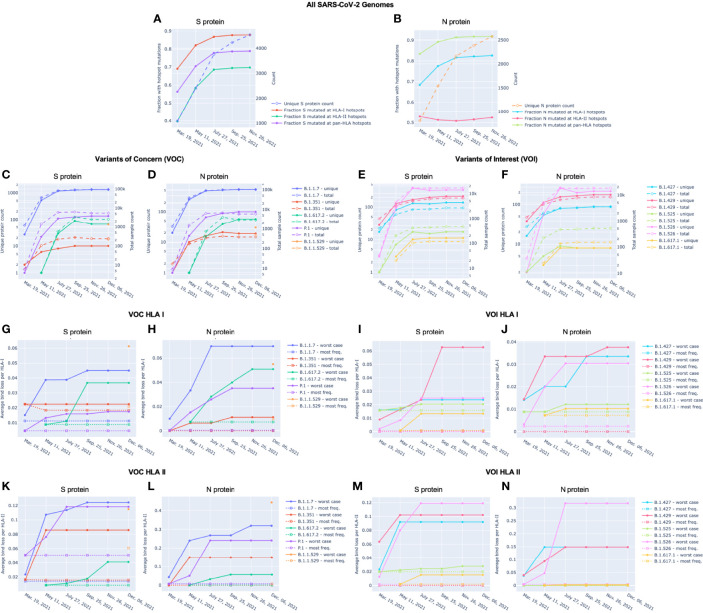
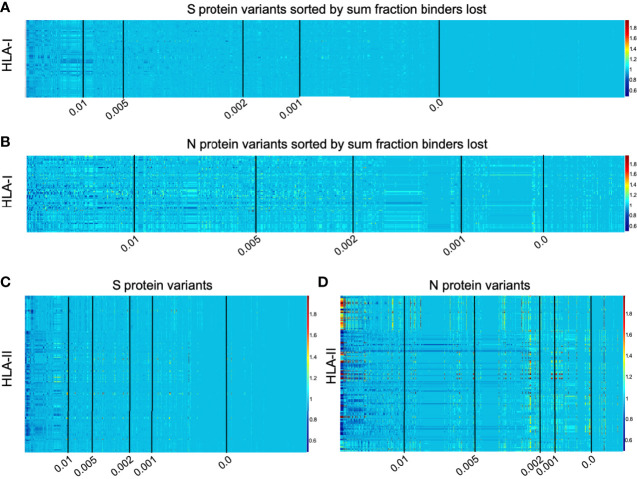
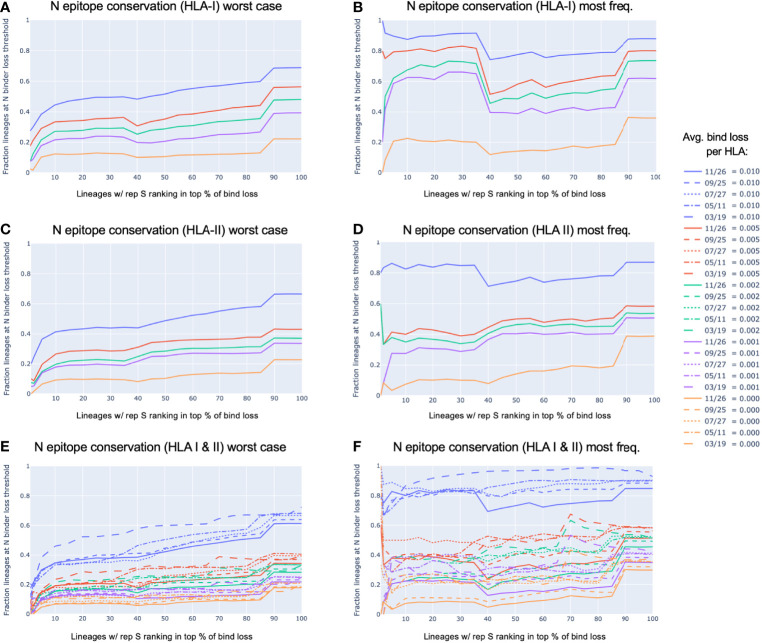
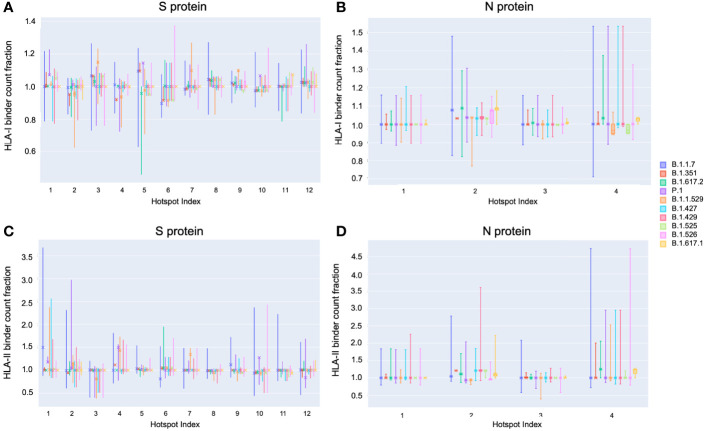
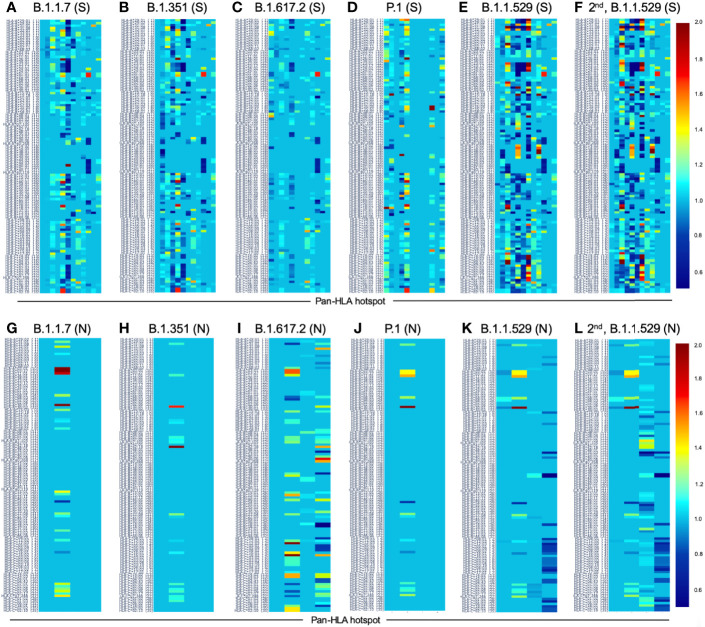
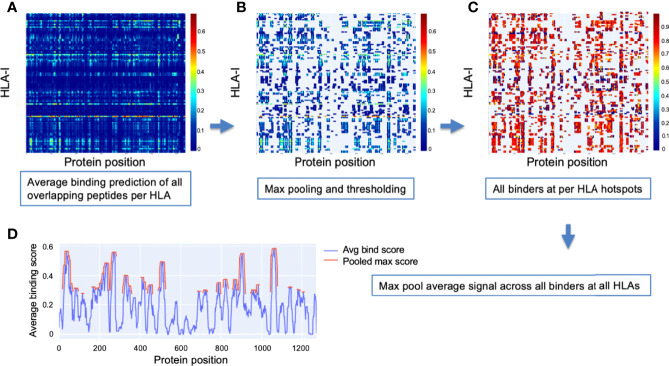
To provide a unique global view of the relative potential for evasion of CD8+ and CD4+ T cells by SARS-CoV-2 lineages as they evolve over time, we performed a comprehensive analysis of predicted HLA-I and HLA-II binding peptides in Spike (S) and Nucleocapsid (N) protein sequences of all available SARS-CoV-2 genomes as provided by NIH NCBI at a bi-monthly interval between March and December of 2021. A data supplement of all B.1.1.529 (Omicron) genomes from GISAID in early December was also used to capture the rapidly spreading variant. A key finding is that throughout continued viral evolution and increasing rates of mutations occurring at T-cell epitope hotspots, protein instances with worst-case binding loss did not become the most frequent for any Variant of Concern (VOC) or Variant of Interest (VOI) lineage; suggesting T-cell evasion is not likely to be a dominant evolutionary pressure on SARS-CoV-2. We also determined that throughout the course of the pandemic in 2021, there remained a relatively steady ratio of viral variants that exhibit conservation of epitopes in the N protein, despite significant potential for epitope loss in S relative to other lineages. We further localized conserved regions in N with high epitope yield potential, and illustrated heterogeneity in HLA-I binding across the S protein consistent with empirical observations. Although Omicron's high volume of mutations caused it to exhibit more epitope loss potential than most frequently observed versions of proteins in almost all other VOCs, epitope candidates across its most frequent N proteins were still largely conserved. This analysis adds to the body of evidence suggesting that N may have merit as an additional antigen to elicit immune responses to vaccination with increased potential to provide sustained protection against COVID-19 disease in the face of emerging variants.
Keywords: HLA; SARS-CoV-2; binding; conservation; epitope; nucleocapsid; spike; variants.
Copyright © 2022 Wnuk, Sudol, Spilman and Soon-Shiong.
Conflict of interest statement
All authors are affiliated with Immunitybio, Inc. a company that is developing SARS-CoV-2 vaccines. The study received funding from ImmunityBio, Inc. The funder designed study, performed all analyses and wrote the submitted manuscript. Proprietary software used for HLA binding predictions will be made commercially available to interested researchers and is covered by the following intellectual property: International Application Number PCT/US2019/046582 (International Publication Number WO 2020/046587 Α2); USPTO Application 17670385.
Figures
Similar articles
-
In silico analysis of mutant epitopes in new SARS-CoV-2 lineages suggest global enhanced CD8+ T cell reactivity and also signs of immune response escape.Infect Genet Evol. 2022 Apr;99:105236. doi: 10.1016/j.meegid.2022.105236. Epub 2022 Feb 8. Infect Genet Evol. 2022. PMID: 35149224 Free PMC article.
-
CD8+ T-Cell Epitope Variations Suggest a Potential Antigen HLA-A2 Binding Deficiency for Spike Protein of SARS-CoV-2.Front Immunol. 2022 Jan 18;12:764949. doi: 10.3389/fimmu.2021.764949. eCollection 2021. Front Immunol. 2022. PMID: 35116022 Free PMC article.
-
COVID-19 coronavirus vaccine T cell epitope prediction analysis based on distributions of HLA class I loci (HLA-A, -B, -C) across global populations.Hum Vaccin Immunother. 2021 Apr 3;17(4):1097-1108. doi: 10.1080/21645515.2020.1823777. Epub 2020 Nov 11. Hum Vaccin Immunother. 2021. PMID: 33175614 Free PMC article.
-
Evolution of SARS-CoV-2-specific CD4+ T cell epitopes.Immunogenetics. 2023 Jun;75(3):283-293. doi: 10.1007/s00251-023-01295-8. Epub 2023 Jan 31. Immunogenetics. 2023. PMID: 36719467 Free PMC article. Review.
-
Degenerate CD8 Epitopes Mapping to Structurally Constrained Regions of the Spike Protein: A T Cell-Based Way-Out From the SARS-CoV-2 Variants Storm.Front Immunol. 2021 Sep 8;12:730051. doi: 10.3389/fimmu.2021.730051. eCollection 2021. Front Immunol. 2021. PMID: 34566990 Free PMC article. Review.
Cited by
-
Overview of immunological & virological factors driving the evolution & global spread of SARS-CoV-2 variants.Indian J Med Res. 2023 Sep;158(3):257-268. doi: 10.4103/ijmr.ijmr_2591_22. Indian J Med Res. 2023. PMID: 37815068 Free PMC article. Review.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous