

Multi-Omics Analysis Reveals a Regulatory Network of ZmCCT During Maize Resistance to Gibberella Stalk Rot at the Early Stage
- PMID: 35812937
- PMCID: PMC9260664
- DOI: 10.3389/fpls.2022.917493
Multi-Omics Analysis Reveals a Regulatory Network of ZmCCT During Maize Resistance to Gibberella Stalk Rot at the Early Stage
Abstract
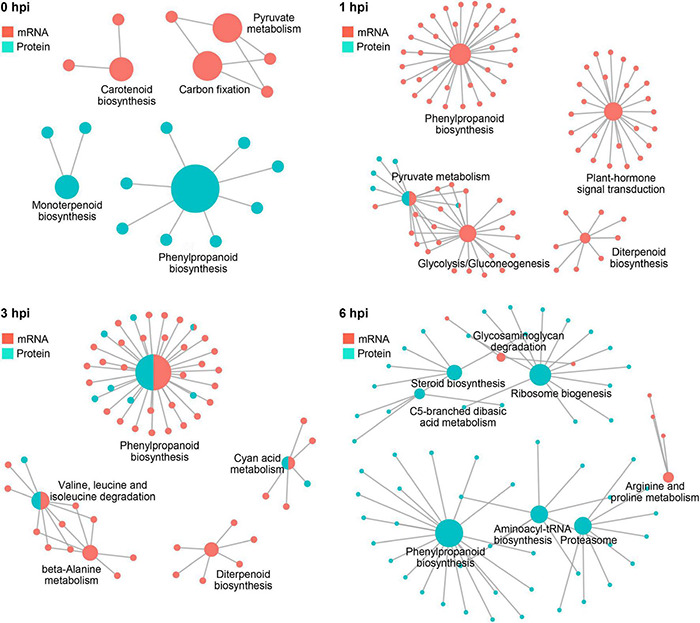
Gibberella stalk rot (GSR) caused by Fusarium graminearum is one of the most devastating diseases in maize; however, the regulatory mechanism of resistance to GSR remains largely unknown. We performed a comparative multi-omics analysis to reveal the early-stage resistance of maize to GSR. We inoculated F. graminearum to the roots of susceptible (Y331) and resistant (Y331-ΔTE) near-isogenic lines containing GSR-resistant gene ZmCCT for multi-omics analysis. Transcriptome detected a rapid reaction that confers resistance at 1-3 hpi as pattern-triggered immunity (PTI) response to GSR. Many key properties were involved in GSR resistance, including genes in photoperiod and hormone pathways of salicylic acid and auxin. The activation of programmed cell death-related genes and a number of metabolic pathways at 6 hpi might be important to prevent further colonization. This is consistent with an integrative analysis of transcriptomics and proteomics that resistant-mediated gene expression reprogramming exhibited a dynamic pattern from 3 to 6 hpi. Further metabolomics analysis revealed that the amount of many chemical compounds was altered in pathways associated with the phenylpropanoid biosynthesis and the phenylalanine metabolism, which may play key roles to confer the GSR resistance. Taken together, we generated a valuable resource to interpret the defense mechanism during early GSR resistance.
Keywords: Fusarium graminearum; gibberella stalk rot; maize disease; metabolomics; multi-omics; plant resistance; transcriptomics.
Copyright © 2022 Tang, Zhang, Zhao, Xu, Wang, Chen and Wang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures









References
-
- Bechtold U., Penfold C. A., Jenkins D. J., Legaie R., Moore J. D., Lawson T., et al. (2016). Time-series transcriptomics reveals that agamous-like22 affects primary metabolism and developmental processes in drought-stressed Arabidopsis. Plant Cell 28 345–366. 10.1105/tpc.15.00910 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources