

Rare KCND3 Loss-of-Function Mutation Associated With the SCA19/22
- PMID: 35813061
- PMCID: PMC9261871
- DOI: 10.3389/fnmol.2022.919199
Rare KCND3 Loss-of-Function Mutation Associated With the SCA19/22
Abstract
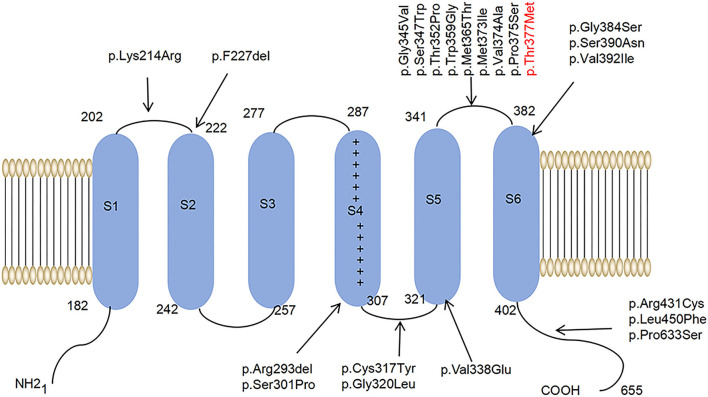
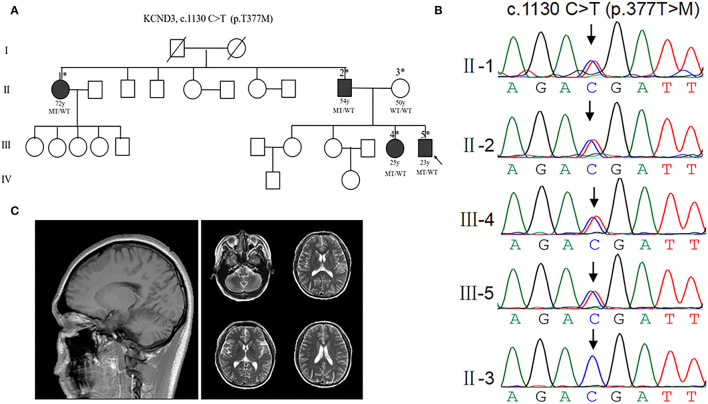
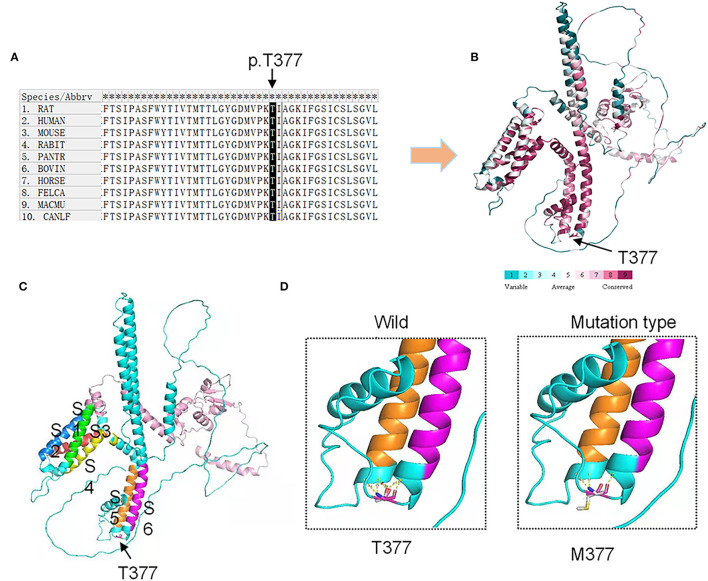
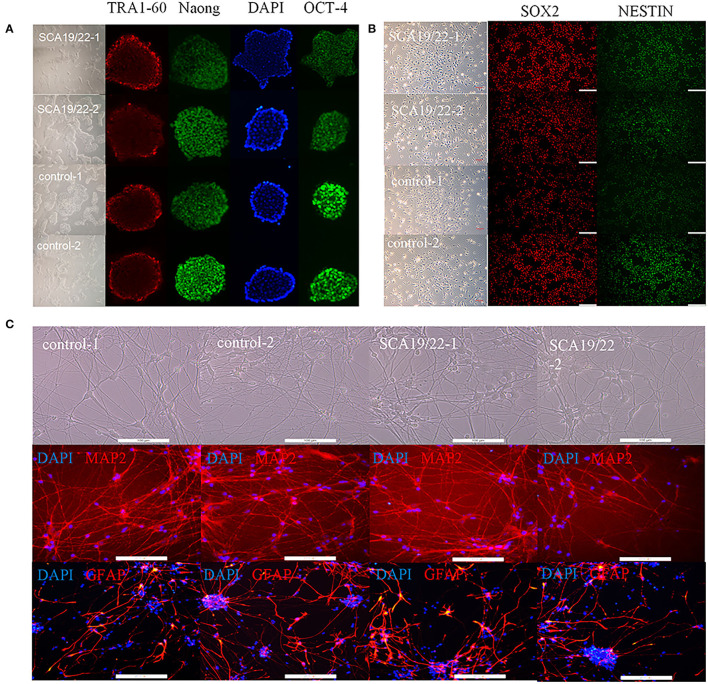
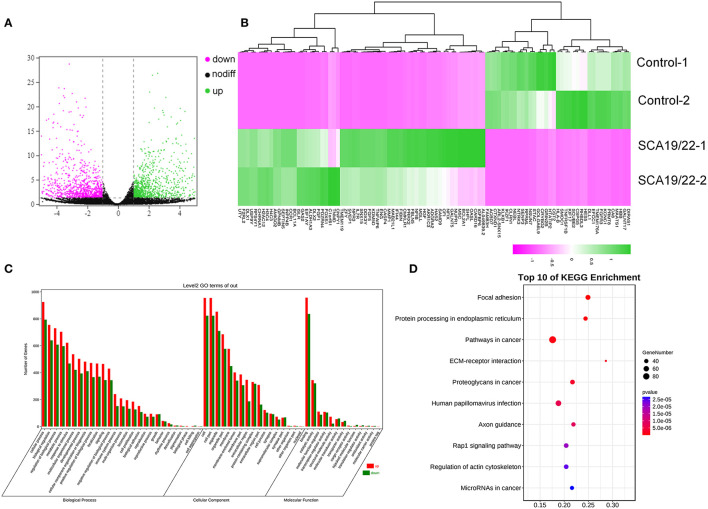
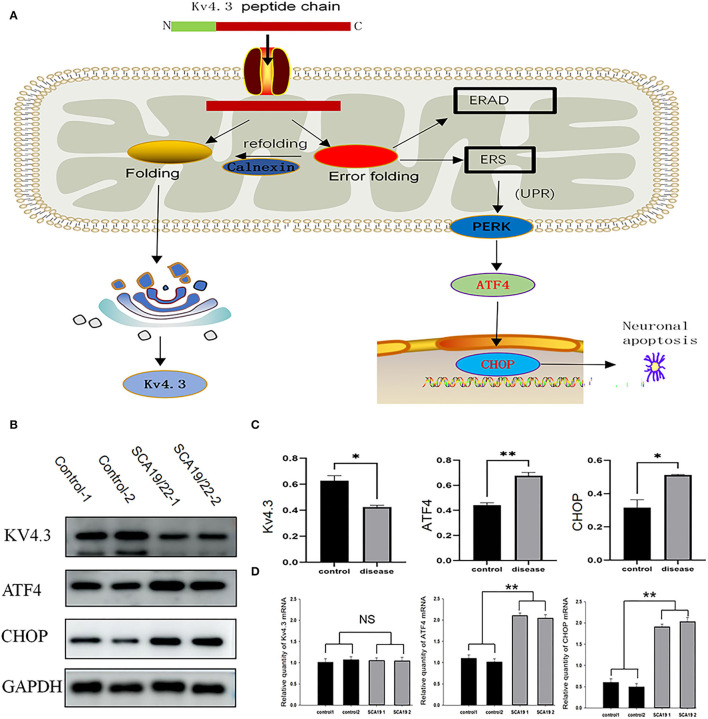
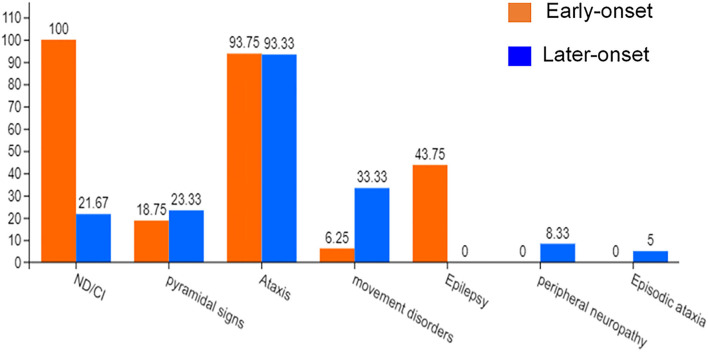
Spinocerebellar ataxia 19/22 (SCA19/22) is a rare neurodegenerative disorder caused by mutations of the KCND3 gene, which encodes the Kv4. 3 protein. Currently, only 22 KCND3 single-nucleotide mutation sites of SCA19/22 have been reported worldwide, and detailed pathogenesis remains unclear. In this study, Sanger sequencing was used to screen 115 probands of cerebellar ataxia families in 67 patients with sporadic cerebellar ataxia and 200 healthy people to identify KCND3 mutations. Mutant gene products showed pathogenicity damage, and the polarity was changed. Next, we established induced pluripotent stem cells (iPSCs) derived from SCA19/22 patients. Using a transcriptome sequencing technique, we found that protein processing in the endoplasmic reticulum was significantly enriched in SCA19/22-iPS-derived neurons and was closely related to endoplasmic reticulum stress (ERS) and apoptosis. In addition, Western blotting of the SCA19/22-iPS-derived neurons showed a reduction in Kv4.3; but, activation of transcription factor 4 (ATF4) and C/EBP homologous protein was increased. Therefore, the c.1130 C>T (p.T377M) mutation of the KCND3 gene may mediate misfold and aggregation of Kv4.3, which activates the ERS and further induces neuron apoptosis involved in SCA19/22.
Keywords: KCND3 mutation; PERK-ATF4-CHOP pathway; SCA19/22; endoplasmic reticulum stress; iPS; neuron; transcriptome (RNA-seq).
Copyright © 2022 Li, Liu, Hao, Fan, Li, Hu, Shi, Fan, Zhang, Ma, Guo, Xu and Shi.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Abisambra J. F., Jinwal U. K., Blair L. J., O'Leary J. C., 3rd, Li Q., Brady S., Wang L., et al. . (2013). Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 33, 9498–9507. 10.1523/JNEUROSCI.5397-12.2013 - DOI - PMC - PubMed
-
- Aimé P., Karuppagounder S. S., Rao A., Chen Y., Burke R. E., Ratan R. R., et al. . (2020). The drug adaptaquin blocks ATF4/CHOP-dependent pro-death Trib3 induction and protects in cellular and mouse models of Parkinson's disease. Neurobiol. Dis. 136, 104725. 10.1016/j.nbd.2019.104725 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Research Materials