

Bacterial Enoyl-Reductases: The Ever-Growing List of Fabs, Their Mechanisms and Inhibition
- PMID: 35814645
- PMCID: PMC9260719
- DOI: 10.3389/fmicb.2022.891610
Bacterial Enoyl-Reductases: The Ever-Growing List of Fabs, Their Mechanisms and Inhibition
Abstract
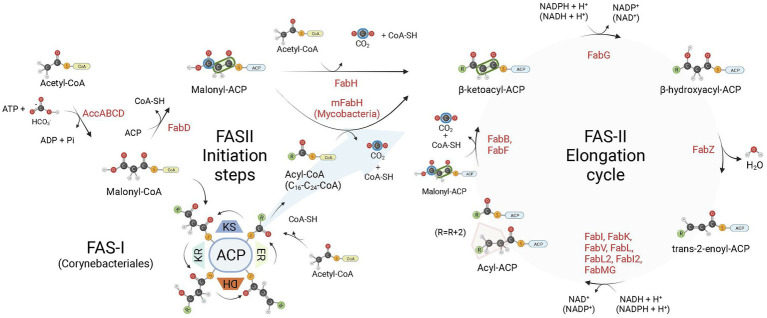
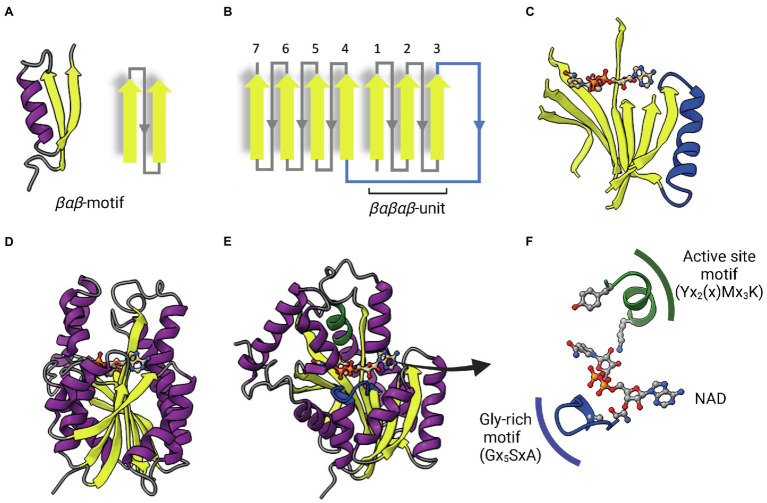
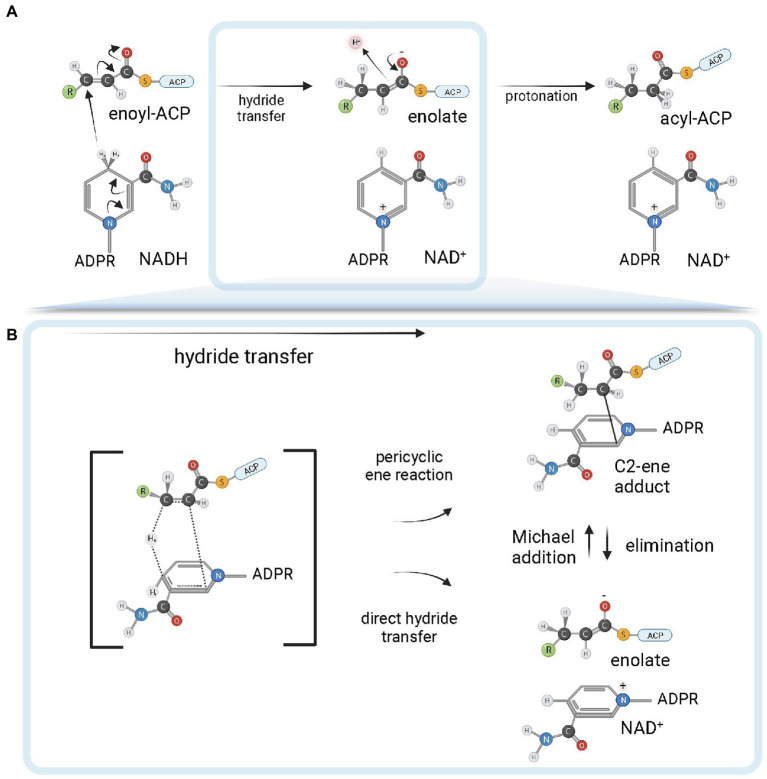
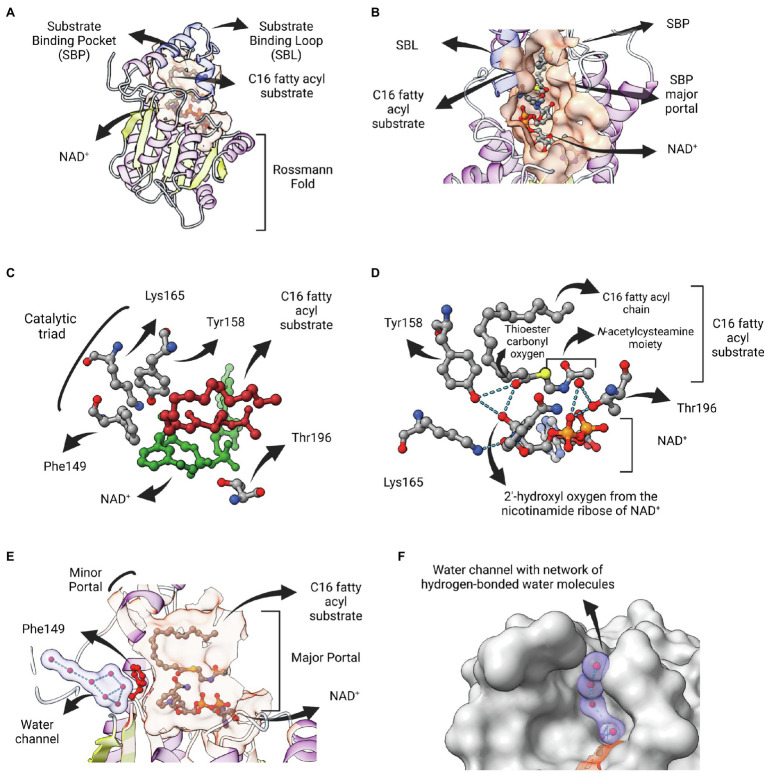
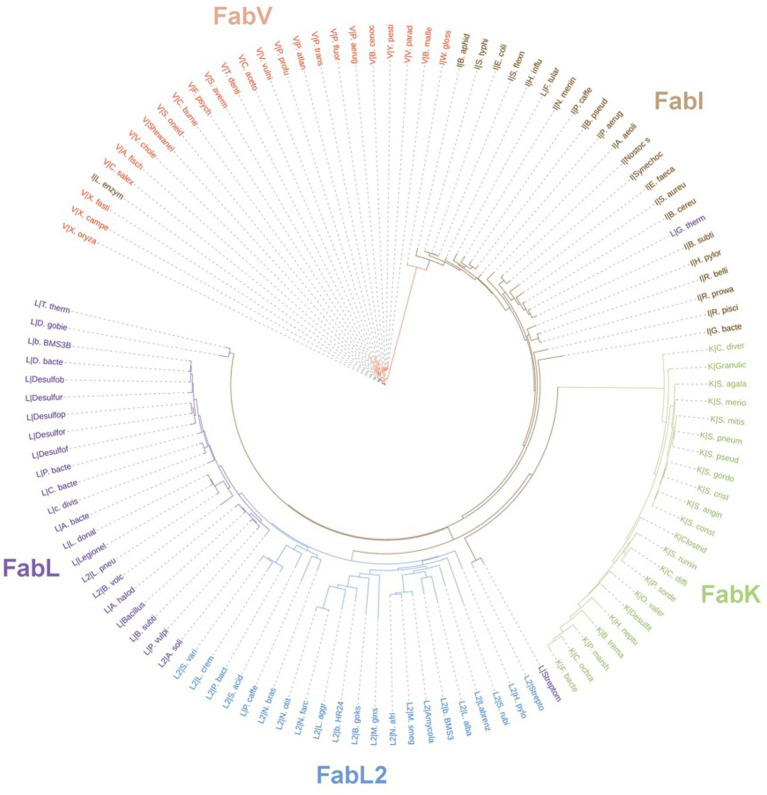
Enoyl-ACP reductases (ENRs) are enzymes that catalyze the last step of the elongation cycle during fatty acid synthesis. In recent years, new bacterial ENR types were discovered, some of them with structures and mechanisms that differ from the canonical bacterial FabI enzymes. Here, we briefly review the diversity of structural and catalytic properties of the canonical FabI and the new FabK, FabV, FabL, and novel ENRs identified in a soil metagenome study. We also highlight recent efforts to use the newly discovered Fabs as targets for drug development and consider the complex evolutionary history of this diverse set of bacterial ENRs.
Keywords: FabI; FabK; FabL2; FabV; Fabl; drug targets; fatty acid synthesis; kinetic mechanisms.
Copyright © 2022 Hopf, Roth, de Souza, Galina, Czeczot, Machado, Basso and Bizarro.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

Similar articles
-
Biochemical and Structural Basis of Triclosan Resistance in a Novel Enoyl-Acyl Carrier Protein Reductase.Antimicrob Agents Chemother. 2018 Jul 27;62(8):e00648-18. doi: 10.1128/AAC.00648-18. Print 2018 Aug. Antimicrob Agents Chemother. 2018. PMID: 29891603 Free PMC article.
-
Vibrio cholerae FabV defines a new class of enoyl-acyl carrier protein reductase.J Biol Chem. 2008 Jan 18;283(3):1308-1316. doi: 10.1074/jbc.M708171200. Epub 2007 Nov 21. J Biol Chem. 2008. PMID: 18032386
-
Biochemical and Structural Insights Concerning Triclosan Resistance in a Novel YX7K Type Enoyl-Acyl Carrier Protein Reductase from Soil Metagenome.Sci Rep. 2019 Oct 28;9(1):15401. doi: 10.1038/s41598-019-51895-2. Sci Rep. 2019. PMID: 31659200 Free PMC article.
-
FabI (enoyl acyl carrier protein reductase) - A potential broad spectrum therapeutic target and its inhibitors.Eur J Med Chem. 2020 Dec 15;208:112757. doi: 10.1016/j.ejmech.2020.112757. Epub 2020 Aug 23. Eur J Med Chem. 2020. PMID: 32883635 Review.
-
Mechanistic diversity and regulation of Type II fatty acid synthesis.Biochem Soc Trans. 2002 Nov;30(Pt 6):1050-5. doi: 10.1042/bst0301050. Biochem Soc Trans. 2002. PMID: 12440970 Review.
Cited by
-
Unsaturated fatty acid synthesis in bacteria: Mechanisms and regulation of canonical and remarkably noncanonical pathways.Biochimie. 2024 Mar;218:137-151. doi: 10.1016/j.biochi.2023.09.007. Epub 2023 Sep 6. Biochimie. 2024. PMID: 37683993 Free PMC article. Review.
-
A Review of Fatty Acid Biosynthesis Enzyme Inhibitors as Promising Antimicrobial Drugs.Pharmaceuticals (Basel). 2023 Mar 10;16(3):425. doi: 10.3390/ph16030425. Pharmaceuticals (Basel). 2023. PMID: 36986522 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources