

Functional landscapes of POLE and POLD1 mutations in checkpoint blockade-dependent antitumor immunity
- PMID: 35817971
- PMCID: PMC10181095
- DOI: 10.1038/s41588-022-01108-w
Functional landscapes of POLE and POLD1 mutations in checkpoint blockade-dependent antitumor immunity
Abstract
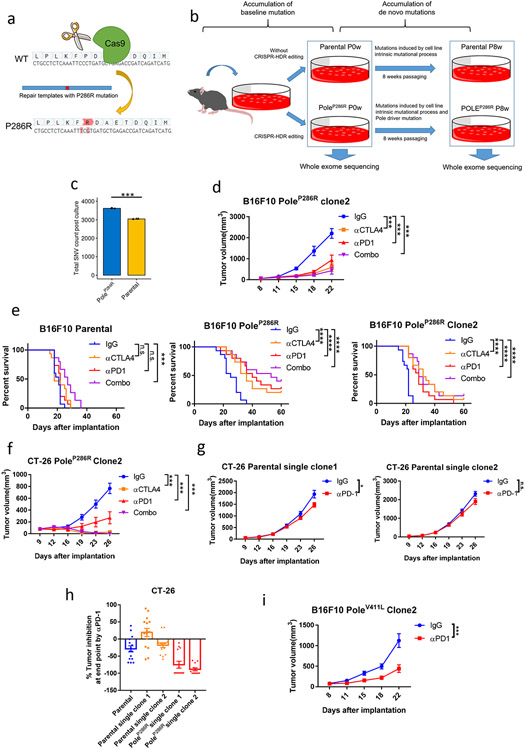
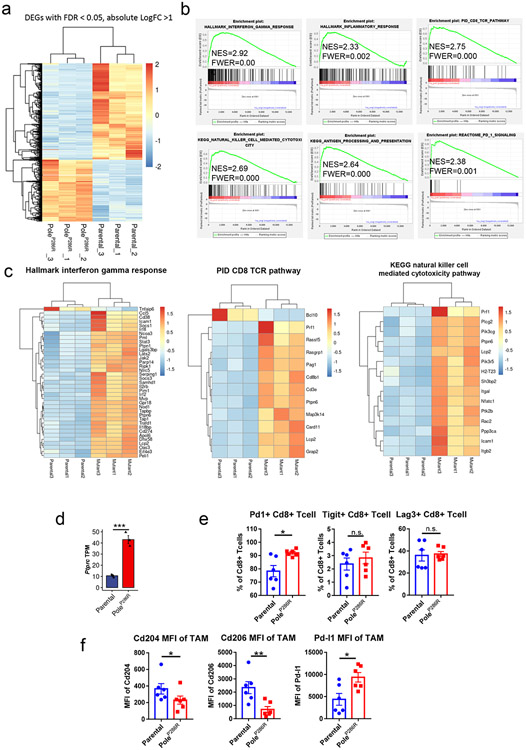
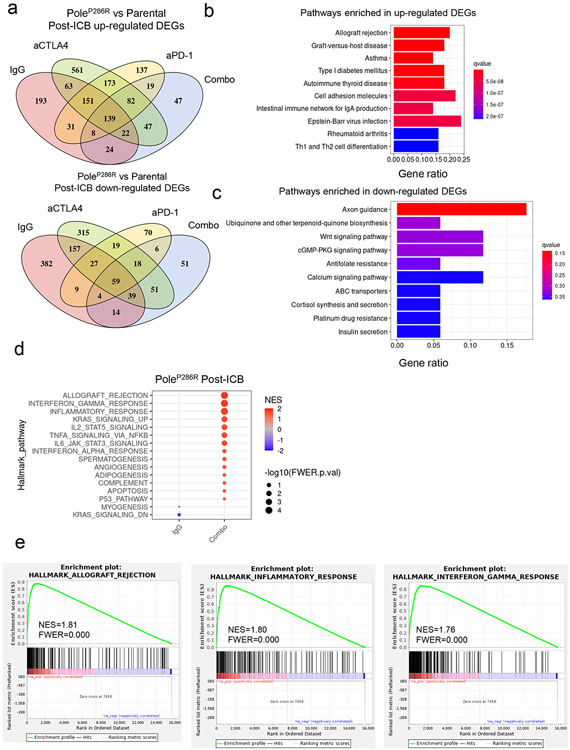
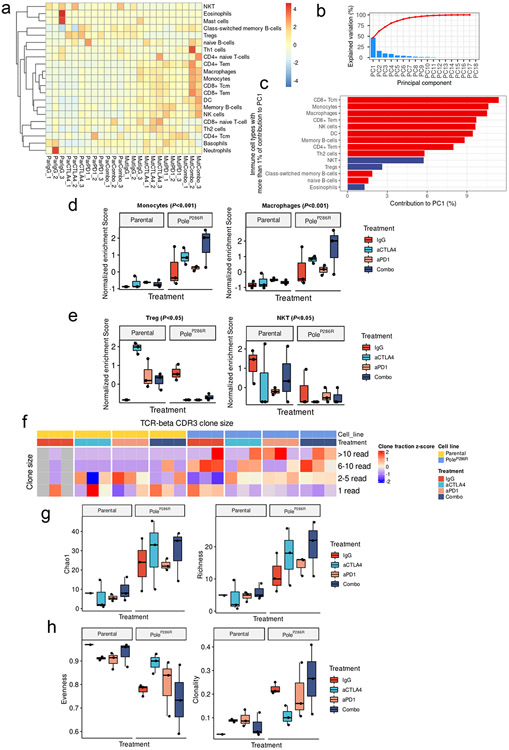
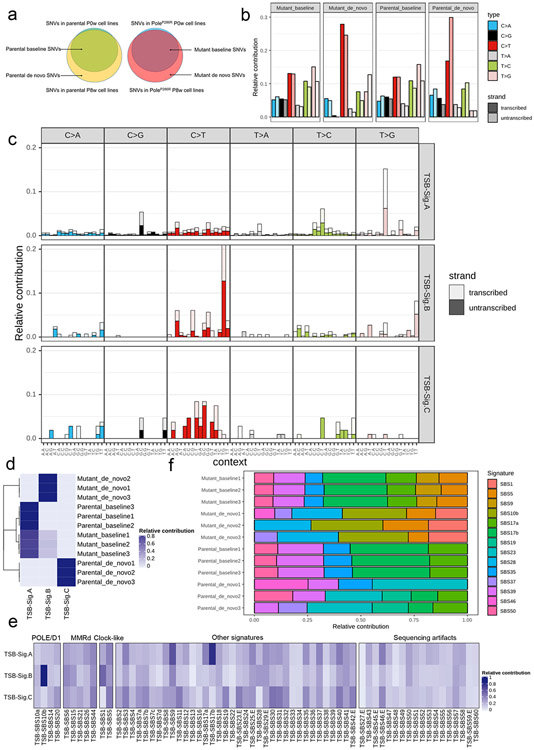
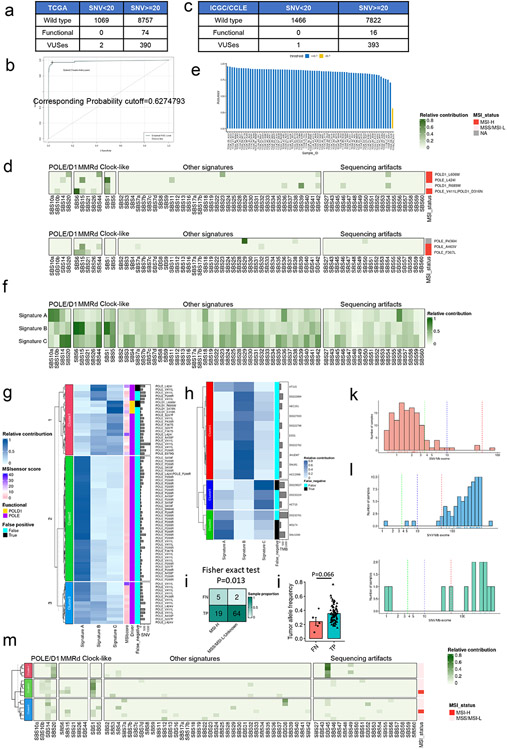
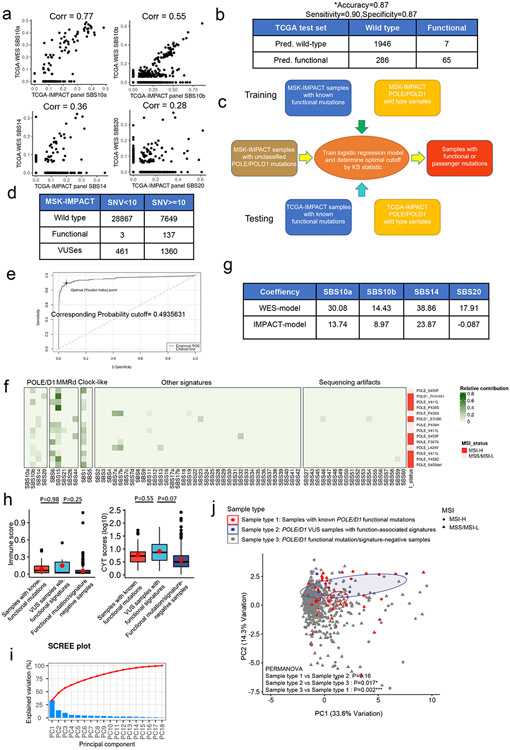
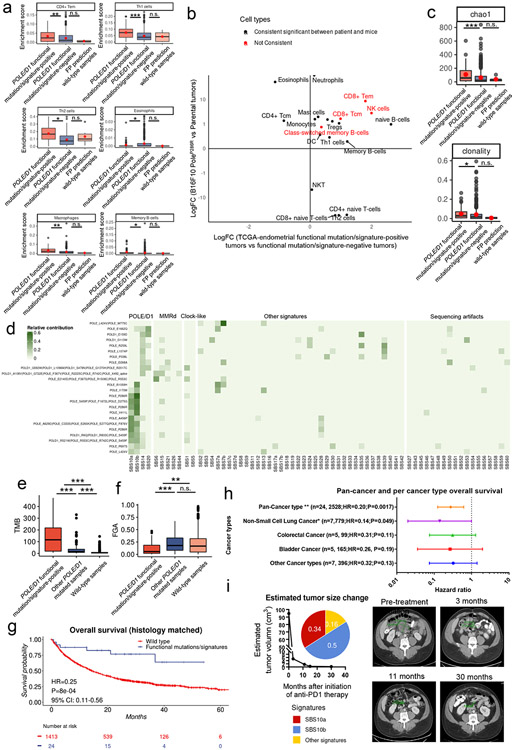
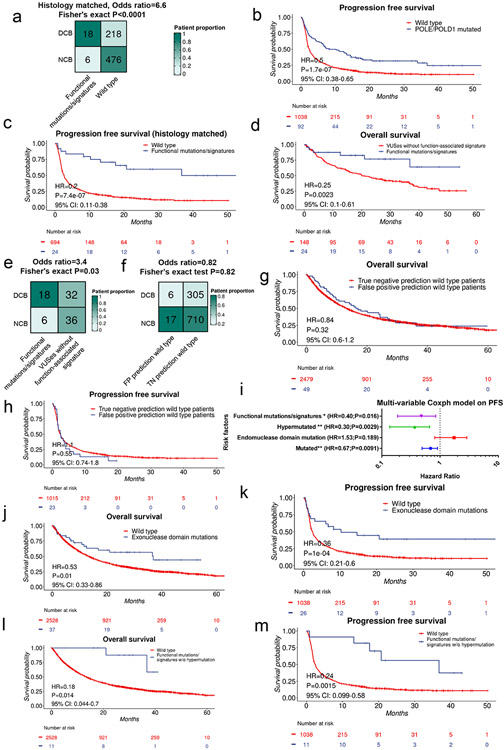
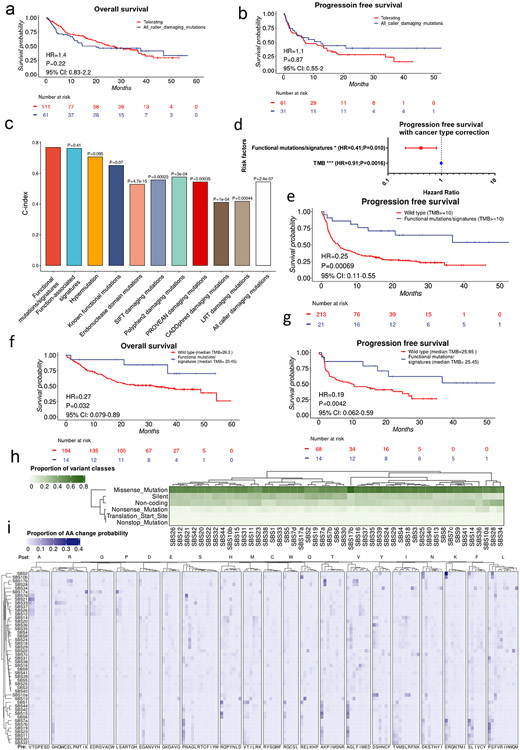
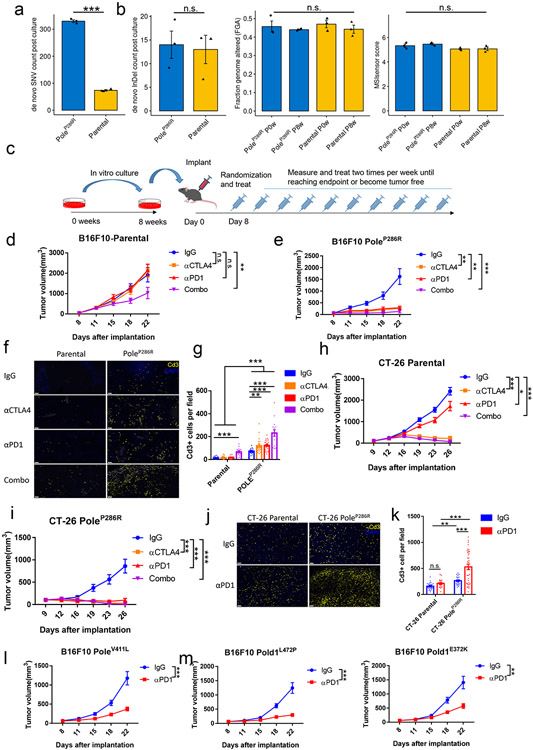
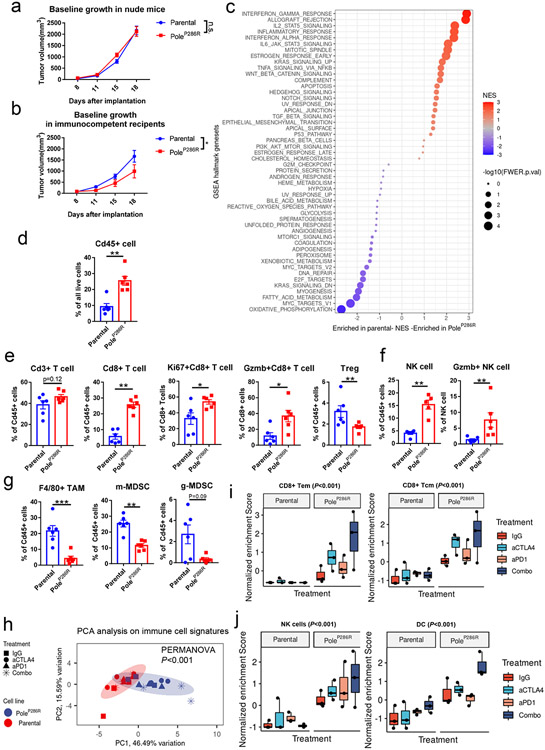
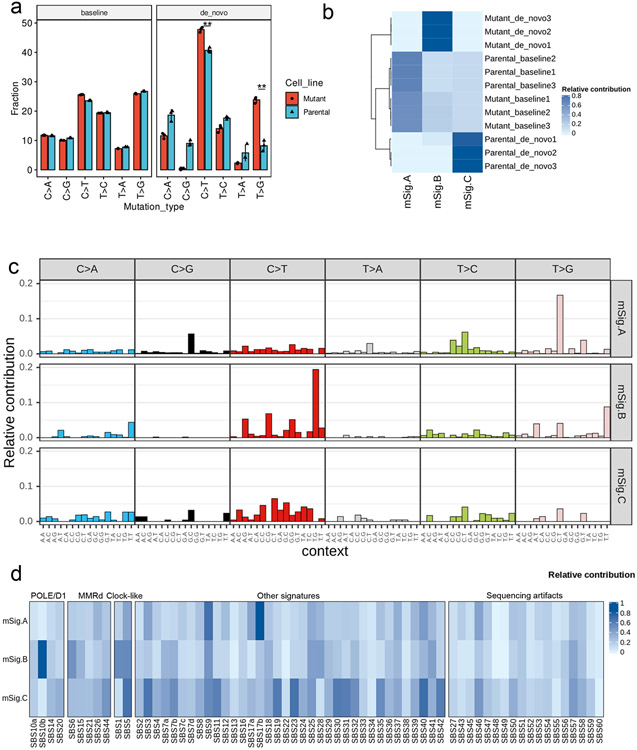
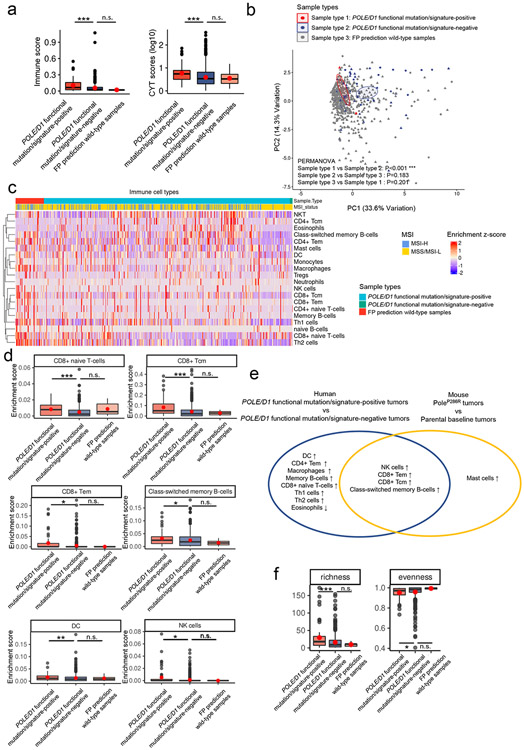
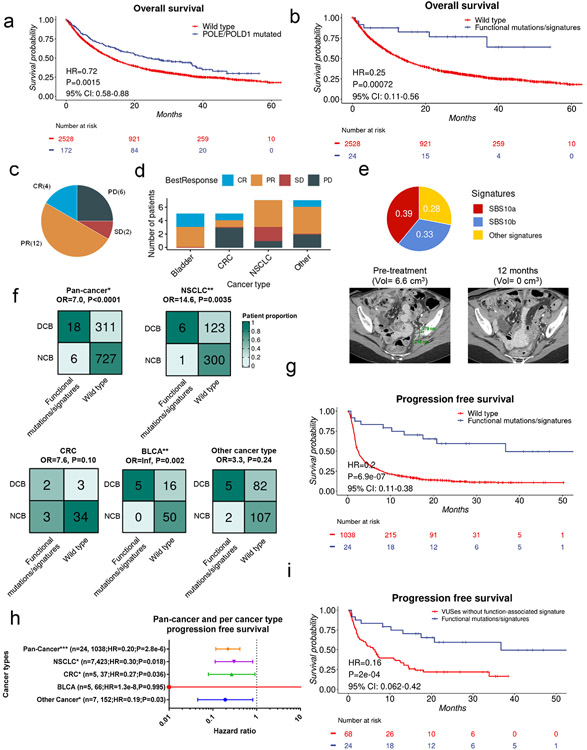
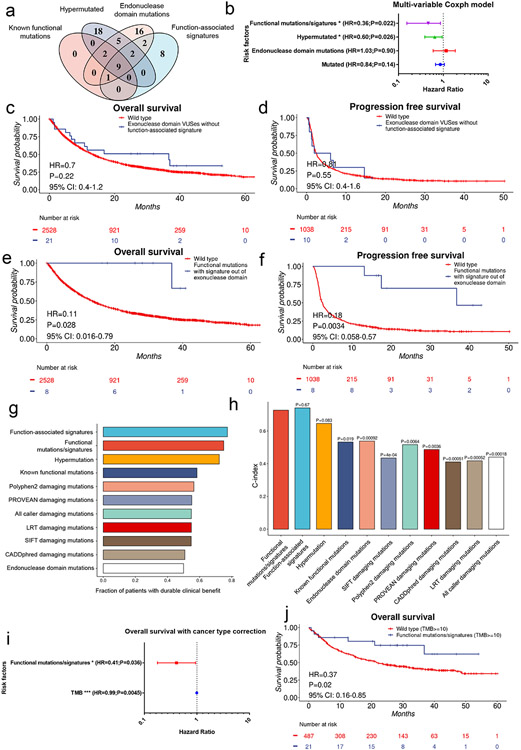
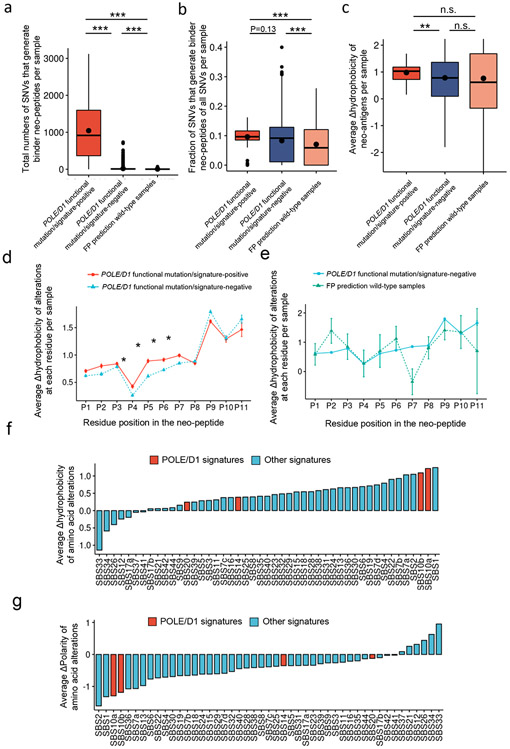
Defects in pathways governing genomic fidelity have been linked to improved response to immune checkpoint blockade therapy (ICB). Pathogenic POLE/POLD1 mutations can cause hypermutation, yet how diverse mutations in POLE/POLD1 influence antitumor immunity following ICB is unclear. Here, we comprehensively determined the effect of POLE/POLD1 mutations in ICB and elucidated the mechanistic impact of these mutations on tumor immunity. Murine syngeneic tumors harboring Pole/Pold1 functional mutations displayed enhanced antitumor immunity and were sensitive to ICB. Patients with POLE/POLD1 mutated tumors harboring telltale mutational signatures respond better to ICB than patients harboring wild-type or signature-negative tumors. A mutant POLE/D1 function-associated signature-based model outperformed several traditional approaches for identifying POLE/POLD1 mutated patients that benefit from ICB. Strikingly, the spectrum of mutational signatures correlates with the biochemical features of neoantigens. Alterations that cause POLE/POLD1 function-associated signatures generate T cell receptor (TCR)-contact residues with increased hydrophobicity, potentially facilitating T cell recognition. Altogether, the functional landscapes of POLE/POLD1 mutations shape immunotherapy efficacy.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
