

A sequence-based global map of regulatory activity for deciphering human genetics
- PMID: 35817977
- PMCID: PMC9279145
- DOI: 10.1038/s41588-022-01102-2
A sequence-based global map of regulatory activity for deciphering human genetics
Abstract
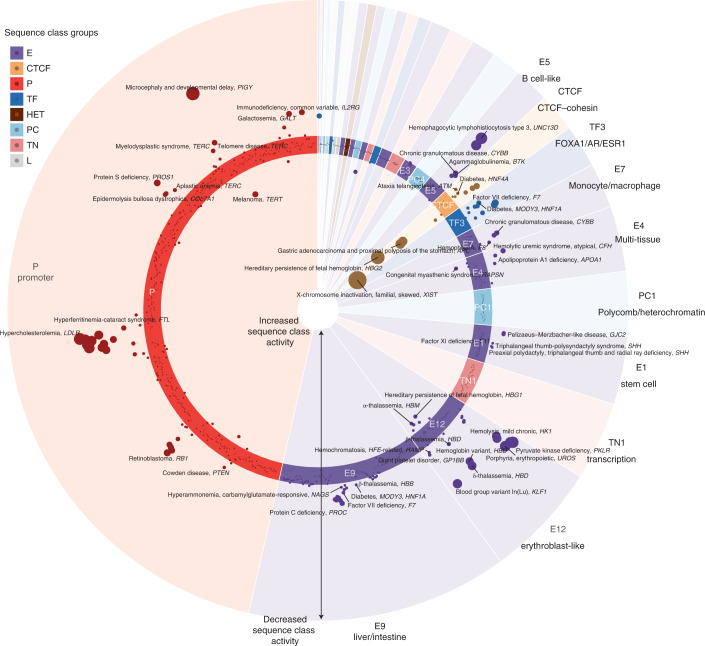
Epigenomic profiling has enabled large-scale identification of regulatory elements, yet we still lack a systematic mapping from any sequence or variant to regulatory activities. We address this challenge with Sei, a framework for integrating human genetics data with sequence information to discover the regulatory basis of traits and diseases. Sei learns a vocabulary of regulatory activities, called sequence classes, using a deep learning model that predicts 21,907 chromatin profiles across >1,300 cell lines and tissues. Sequence classes provide a global classification and quantification of sequence and variant effects based on diverse regulatory activities, such as cell type-specific enhancer functions. These predictions are supported by tissue-specific expression, expression quantitative trait loci and evolutionary constraint data. Furthermore, sequence classes enable characterization of the tissue-specific, regulatory architecture of complex traits and generate mechanistic hypotheses for individual regulatory pathogenic mutations. We provide Sei as a resource to elucidate the regulatory basis of human health and disease.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
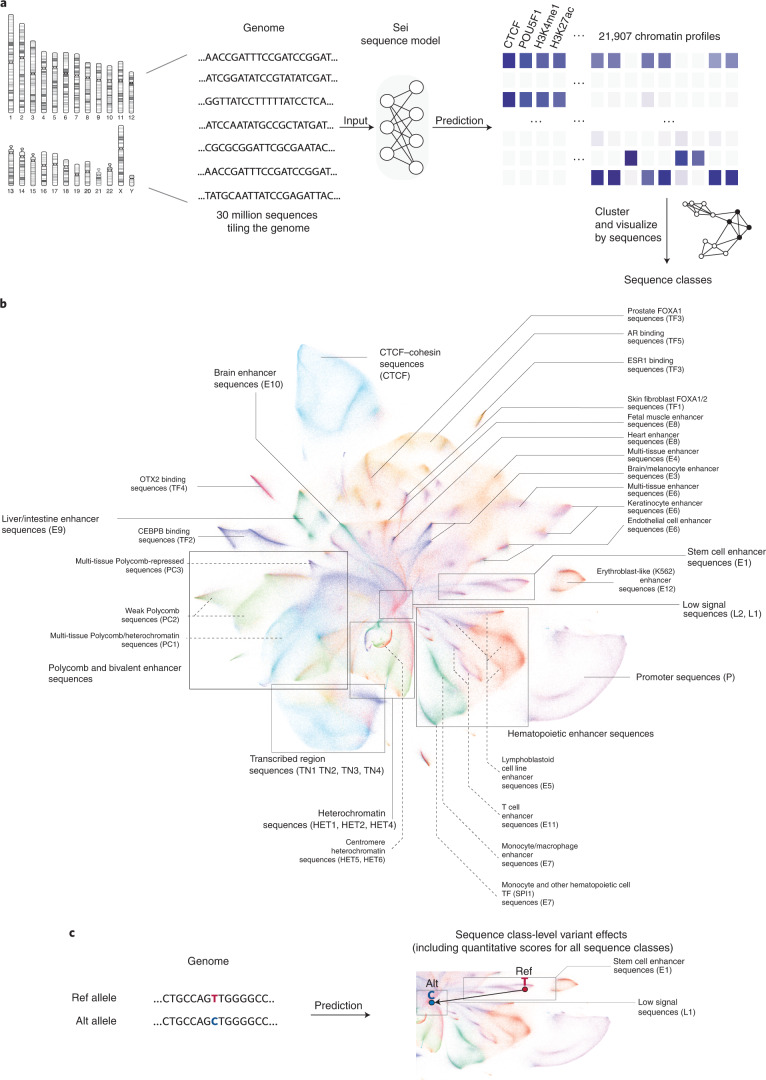
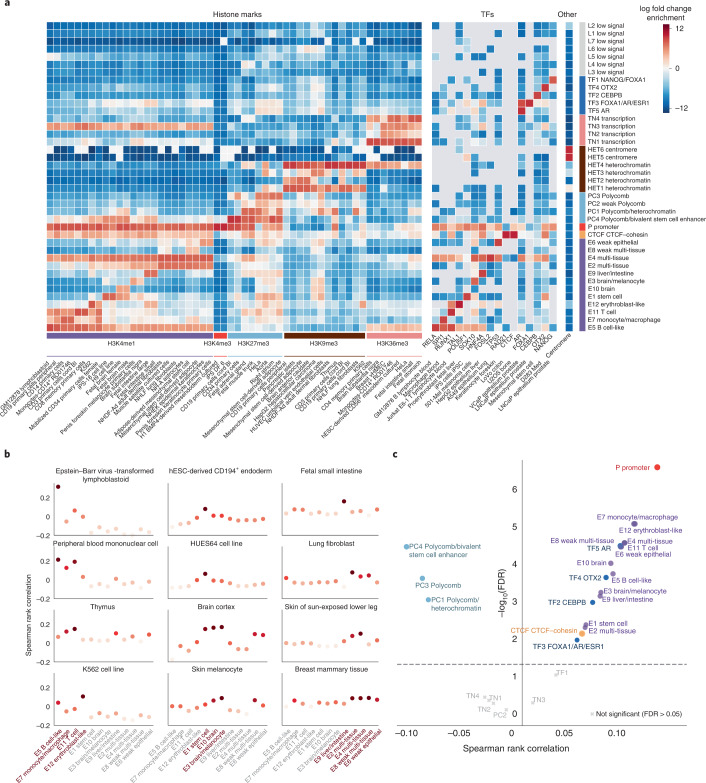
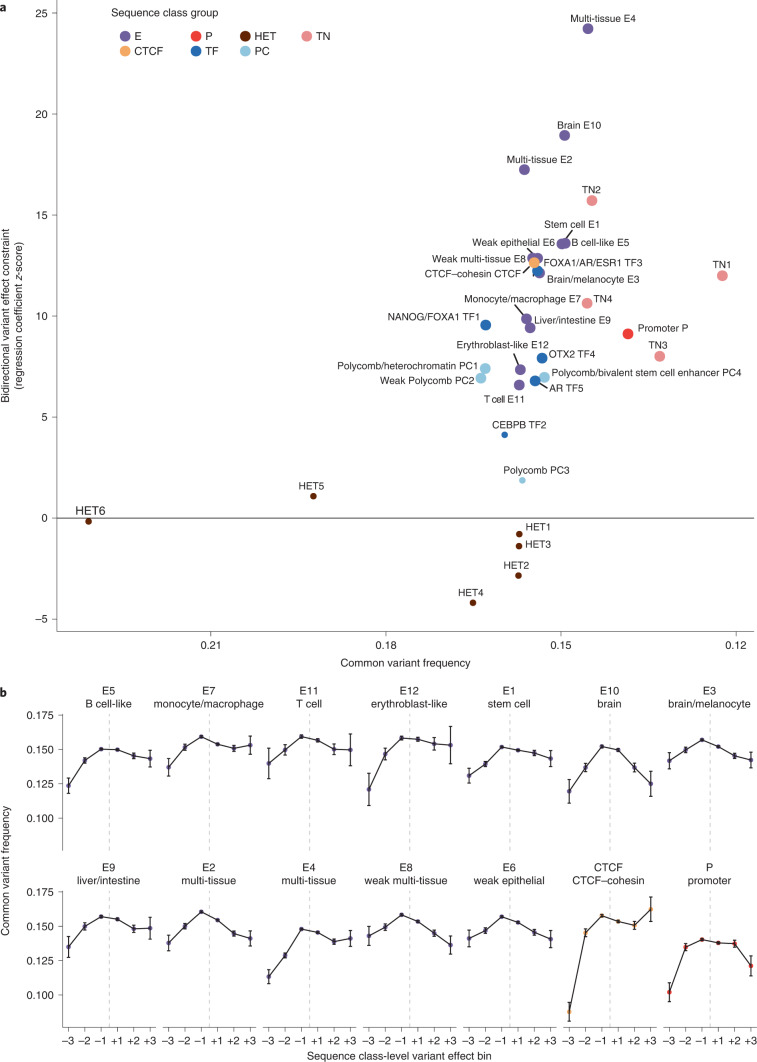
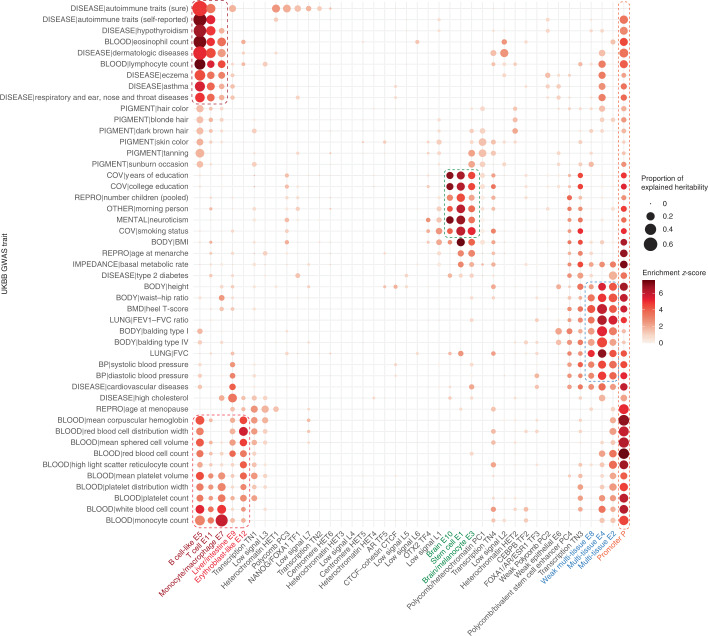
Comment in
-
Automated sequence-based annotation and interpretation of the human genome.Nat Genet. 2022 Jul;54(7):916-917. doi: 10.1038/s41588-022-01123-x. Nat Genet. 2022. PMID: 35817978 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
