

SMYD3 Impedes Small Cell Lung Cancer Sensitivity to Alkylation Damage through RNF113A Methylation-Phosphorylation Cross-talk
- PMID: 35819319
- PMCID: PMC9437563
- DOI: 10.1158/2159-8290.CD-21-0205
SMYD3 Impedes Small Cell Lung Cancer Sensitivity to Alkylation Damage through RNF113A Methylation-Phosphorylation Cross-talk
Abstract
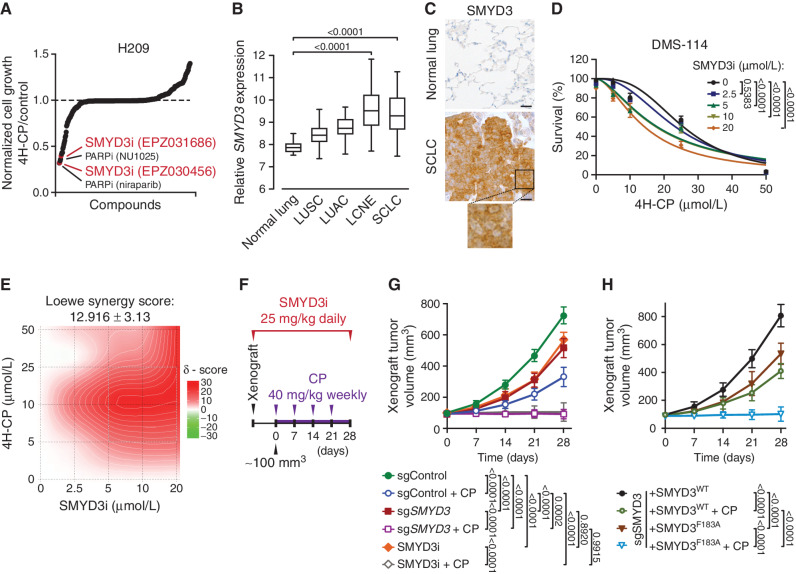
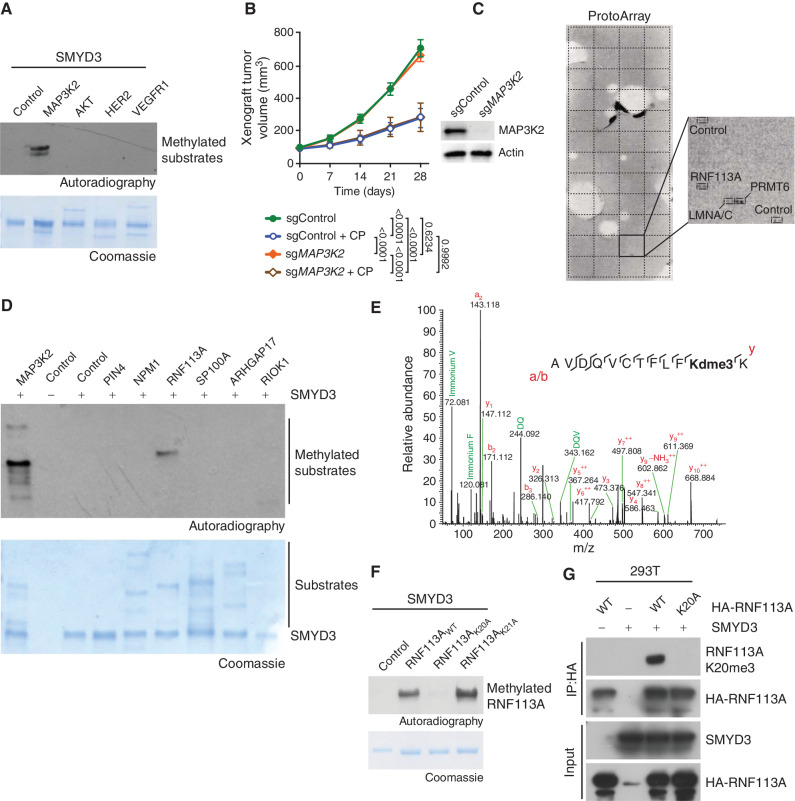
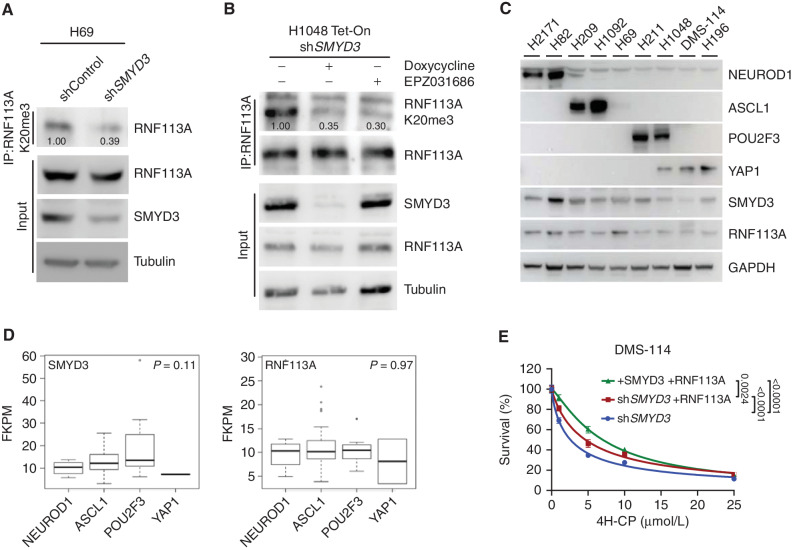
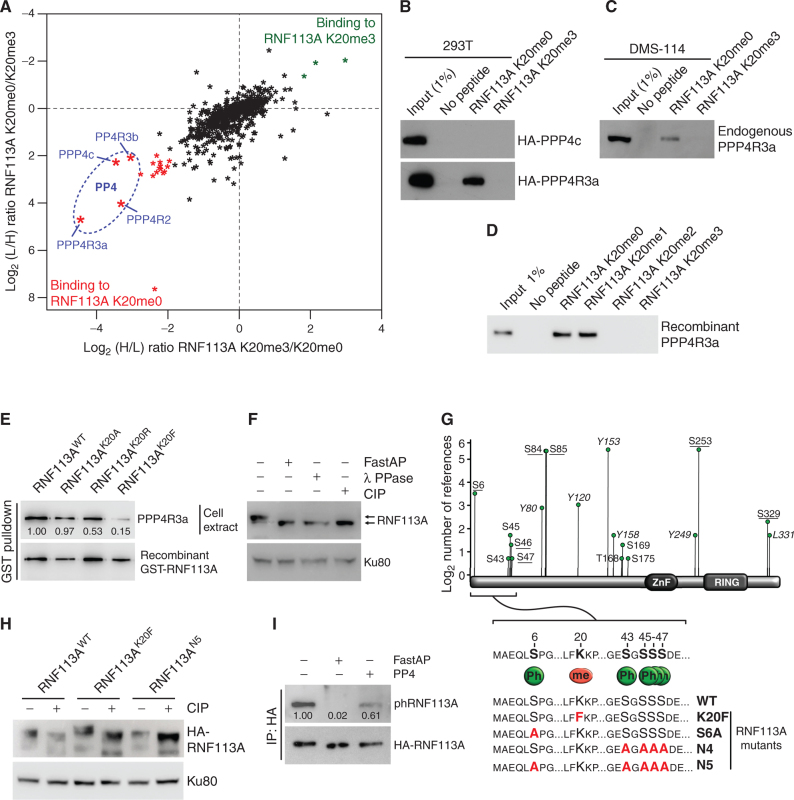
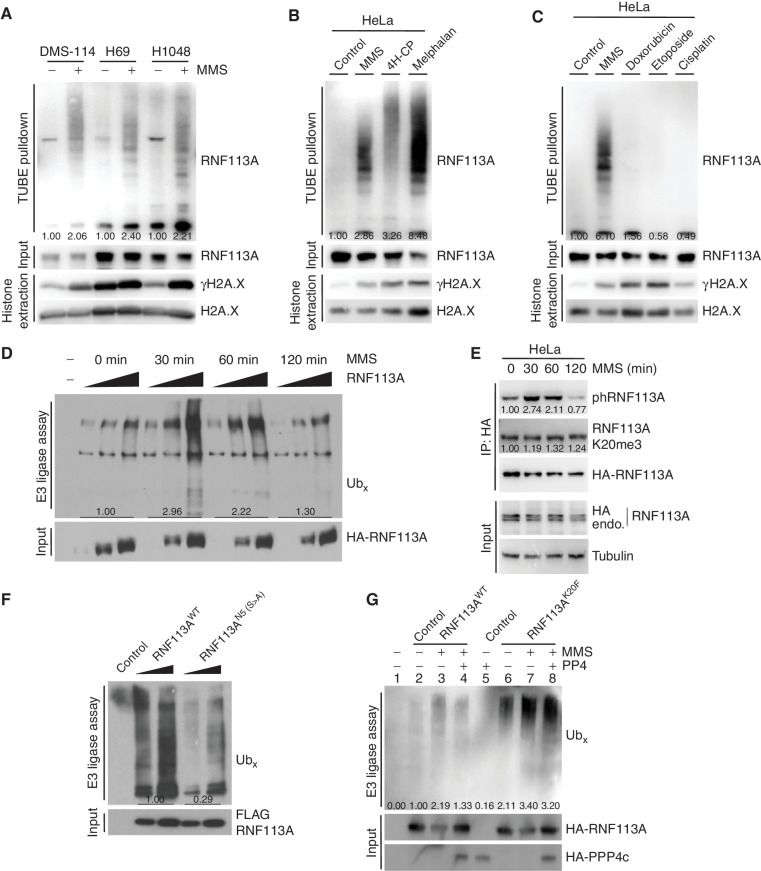
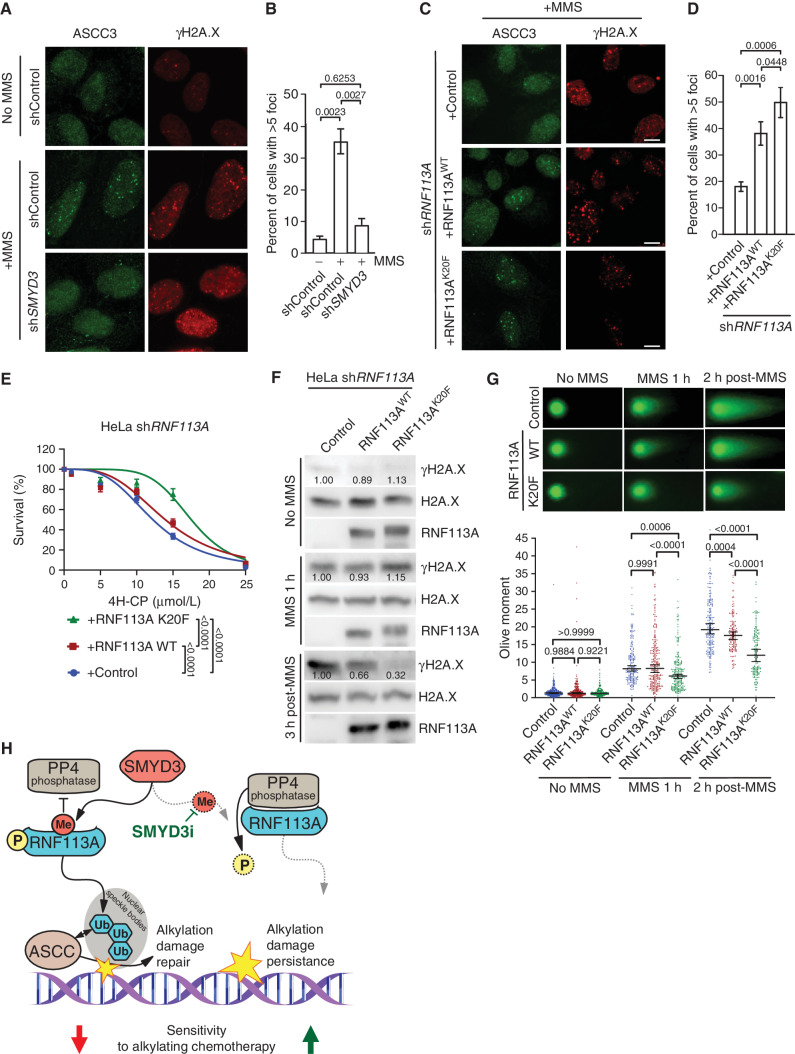
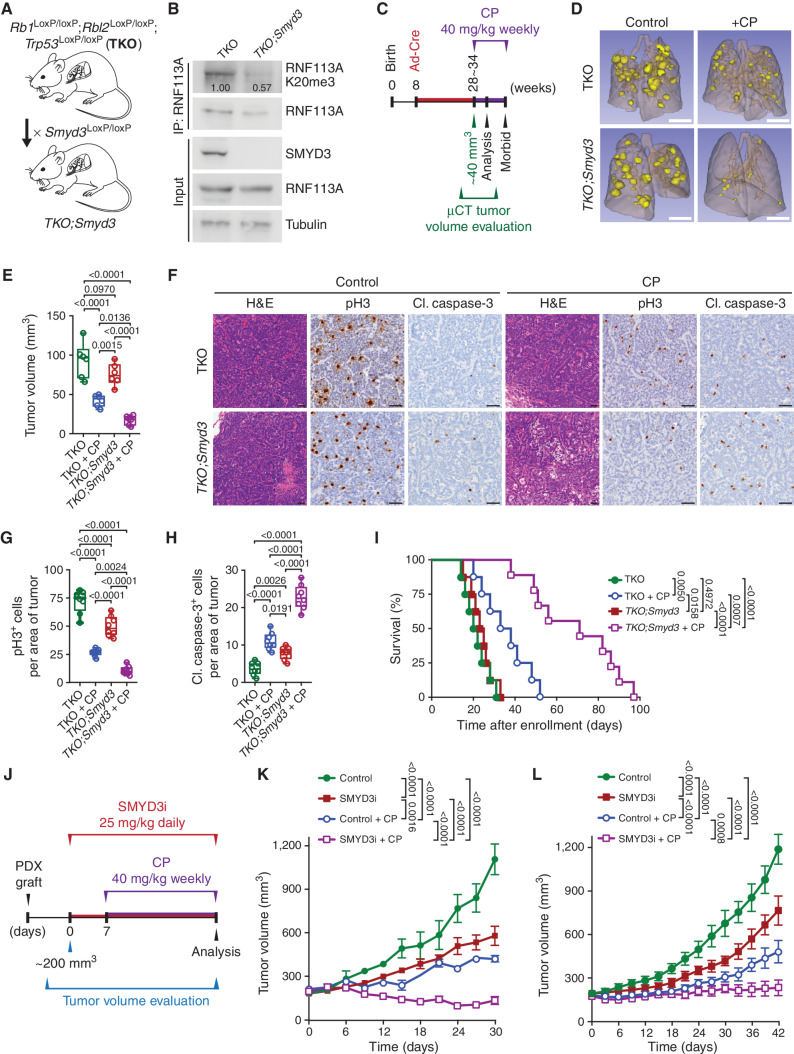
Small cell lung cancer (SCLC) is the most fatal form of lung cancer, with dismal survival, limited therapeutic options, and rapid development of chemoresistance. We identified the lysine methyltransferase SMYD3 as a major regulator of SCLC sensitivity to alkylation-based chemotherapy. RNF113A methylation by SMYD3 impairs its interaction with the phosphatase PP4, controlling its phosphorylation levels. This cross-talk between posttranslational modifications acts as a key switch in promoting and maintaining RNF113A E3 ligase activity, essential for its role in alkylation damage response. In turn, SMYD3 inhibition restores SCLC vulnerability to alkylating chemotherapy. Our study sheds light on a novel role of SMYD3 in cancer, uncovering this enzyme as a mediator of alkylation damage sensitivity and providing a rationale for small-molecule SMYD3 inhibition to improve responses to established chemotherapy.
Significance: SCLC rapidly becomes resistant to conventional chemotherapy, leaving patients with no alternative treatment options. Our data demonstrate that SMYD3 upregulation and RNF113A methylation in SCLC are key mechanisms that control the alkylation damage response. Notably, SMYD3 inhibition sensitizes cells to alkylating agents and promotes sustained SCLC response to chemotherapy. This article is highlighted in the In This Issue feature, p. 2007.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
Comment in
References
-
- Grunberg SM. Cyclophosphamide and etoposide for non-small cell and small cell lung cancer. Drugs 1999;58:11–5. - PubMed
-
- Pujol JL, Daurès JP, Rivière A, Quoix E, Westeel V, Quantin X, et al. Etoposide plus cisplatin with or without the combination of 4′-epidoxorubicin plus cyclophosphamide in treatment of extensive small-cell lung cancer: a French Federation of Cancer Institutes multicenter phase III randomized study. J Natl Cancer Inst 2001;93:300–8. - PubMed
-
- Jiang L, Yang KH, Guan QL, Mi DH, Wang J. Cisplatin plus etoposide versus other platin-based regimens for patients with extensive small-cell lung cancer: a systematic review and meta-analysis of randomised, controlled trials. Intern Med J 2012;42:1297–309. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
