

Genome-wide DNA hypermethylation opposes healing in patients with chronic wounds by impairing epithelial-mesenchymal transition
- PMID: 35819852
- PMCID: PMC9433101
- DOI: 10.1172/JCI157279
Genome-wide DNA hypermethylation opposes healing in patients with chronic wounds by impairing epithelial-mesenchymal transition
Abstract
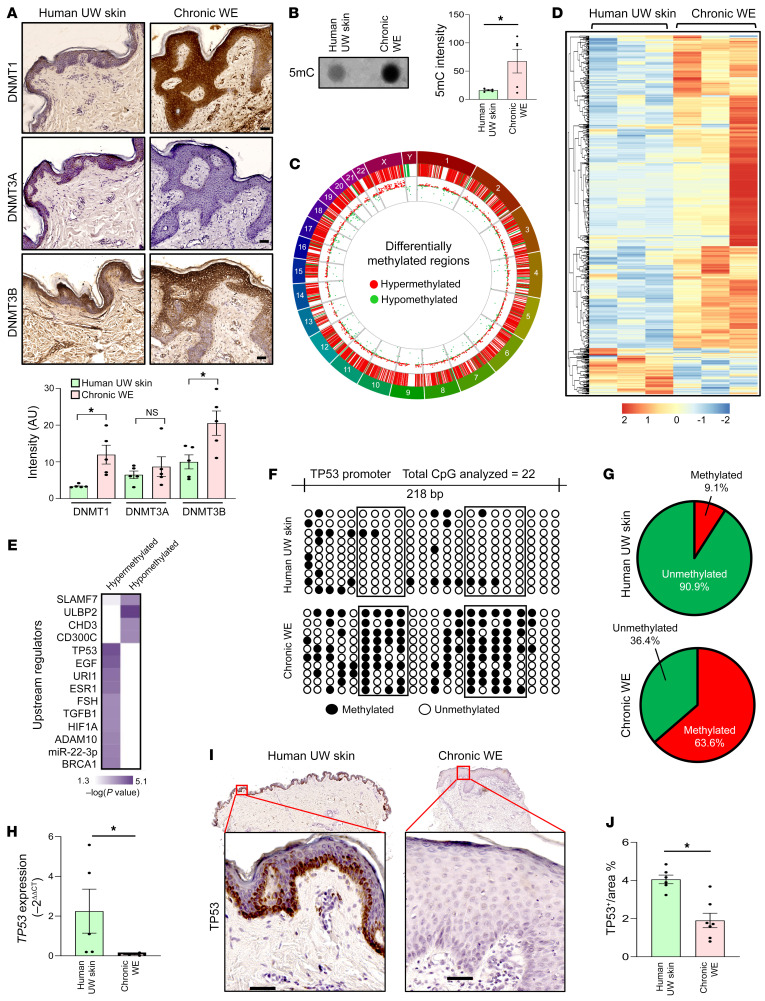
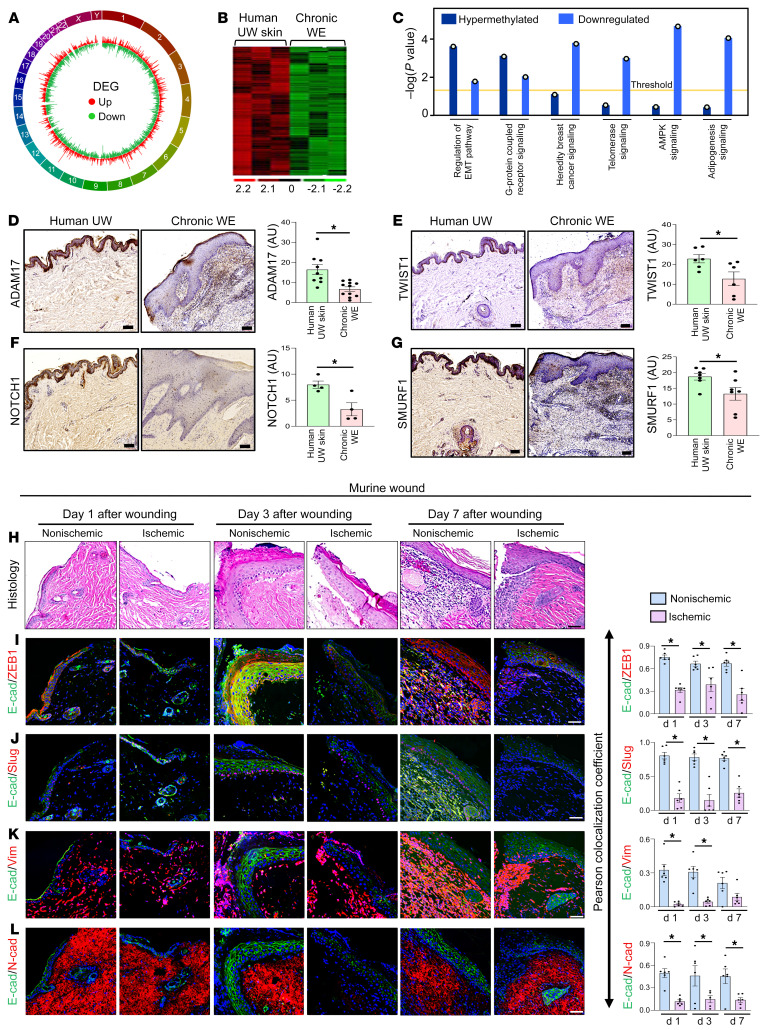
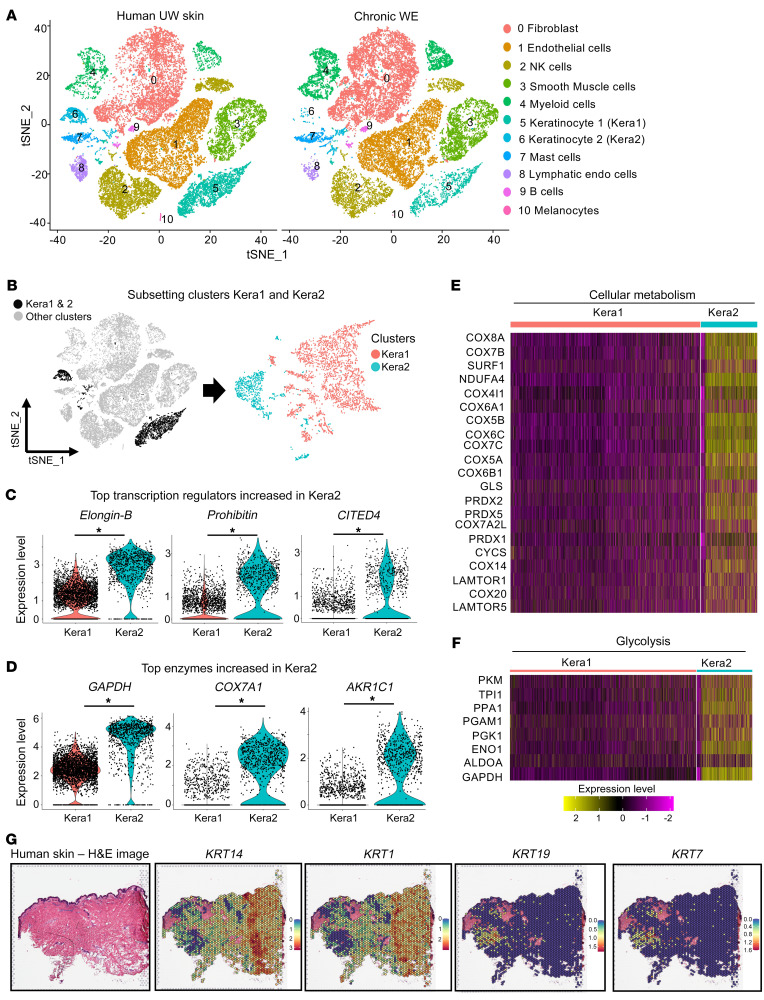
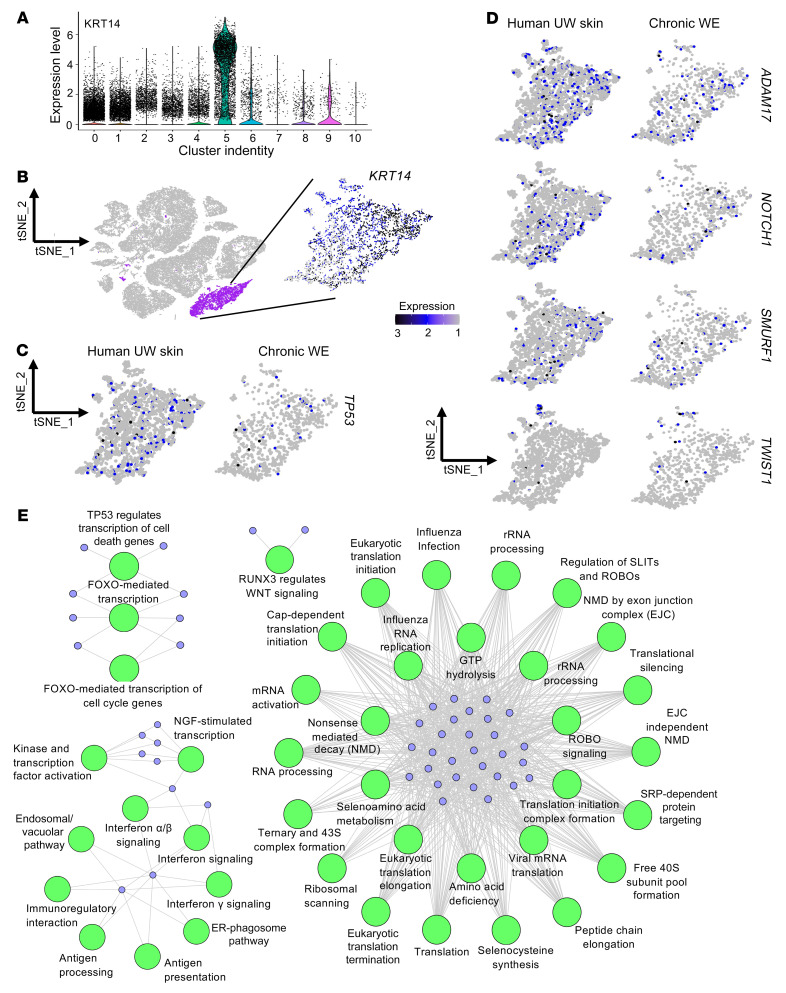
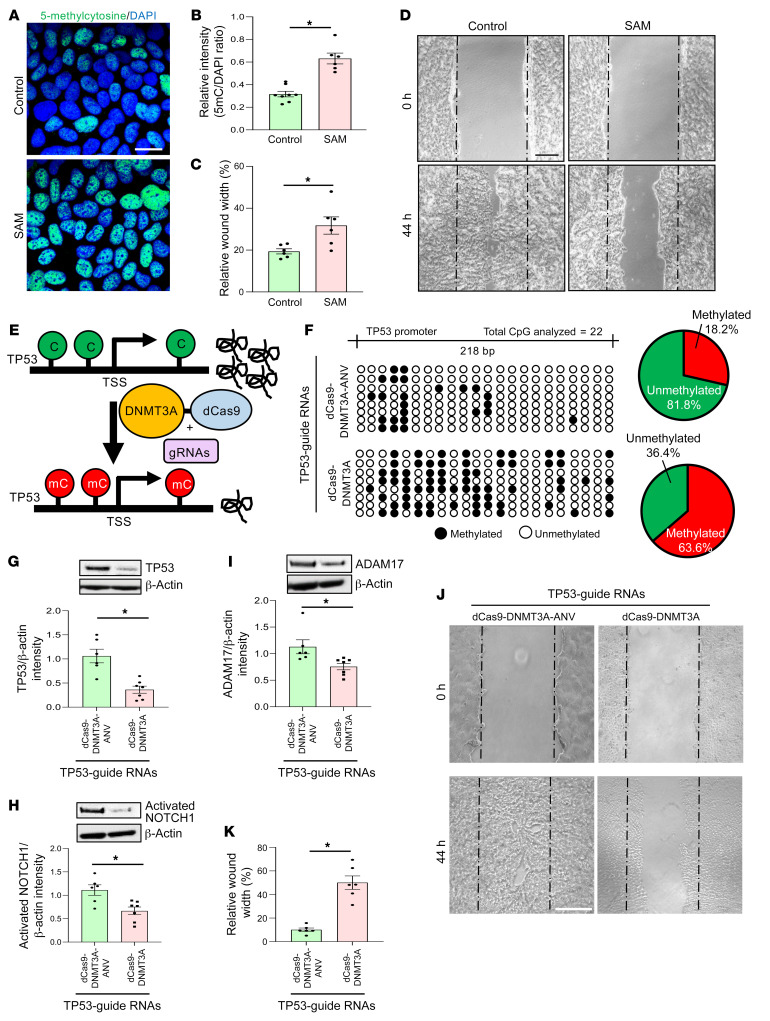
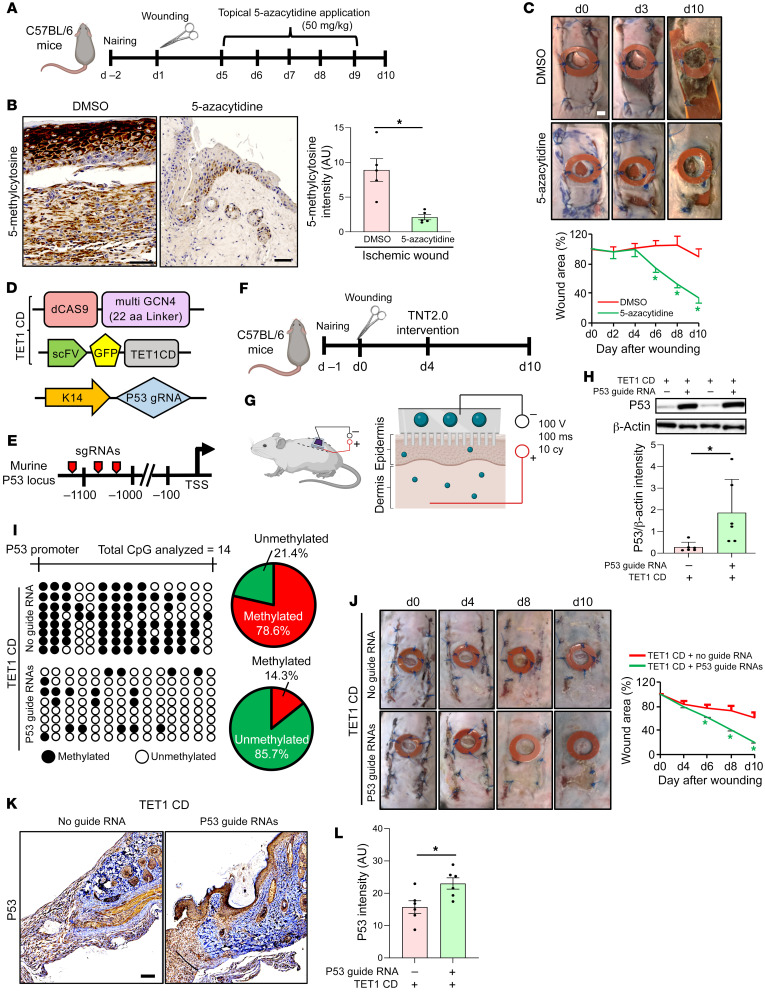
An extreme chronic wound tissue microenvironment causes epigenetic gene silencing. An unbiased whole-genome methylome was studied in the wound-edge tissue of patients with chronic wounds. A total of 4,689 differentially methylated regions (DMRs) were identified in chronic wound-edge skin compared with unwounded human skin. Hypermethylation was more frequently observed (3,661 DMRs) in the chronic wound-edge tissue compared with hypomethylation (1,028 DMRs). Twenty-six hypermethylated DMRs were involved in epithelial-mesenchymal transition (EMT). Bisulfite sequencing validated hypermethylation of a predicted specific upstream regulator TP53. RNA-Seq analysis was performed to qualify findings from methylome analysis. Analysis of the downregulated genes identified the TP53 signaling pathway as being significantly silenced. Direct comparison of hypermethylation and downregulated genes identified 4 genes, ADAM17, NOTCH, TWIST1, and SMURF1, that functionally represent the EMT pathway. Single-cell RNA-Seq studies revealed that these effects on gene expression were limited to the keratinocyte cell compartment. Experimental murine studies established that tissue ischemia potently induces wound-edge gene methylation and that 5'-azacytidine, inhibitor of methylation, improved wound closure. To specifically address the significance of TP53 methylation, keratinocyte-specific editing of TP53 methylation at the wound edge was achieved by a tissue nanotransfection-based CRISPR/dCas9 approach. This work identified that reversal of methylation-dependent keratinocyte gene silencing represents a productive therapeutic strategy to improve wound closure.
Keywords: Dermatology; Epigenetics; Molecular biology; Therapeutics.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
