

Multi-omics profiling of collagen-induced arthritis mouse model reveals early metabolic dysregulation via SIRT1 axis
- PMID: 35821263
- PMCID: PMC9276706
- DOI: 10.1038/s41598-022-16005-9
Multi-omics profiling of collagen-induced arthritis mouse model reveals early metabolic dysregulation via SIRT1 axis
Abstract
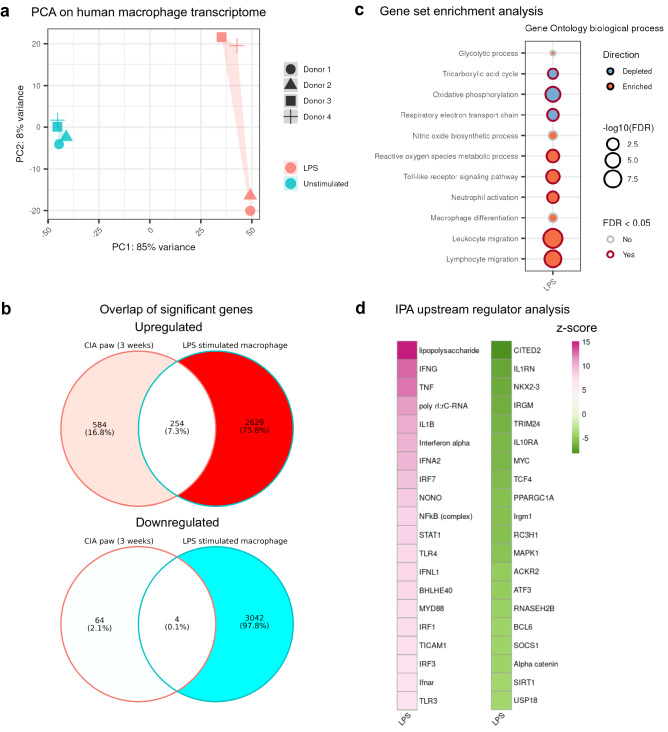
Rheumatoid arthritis (RA) is characterized by joint infiltration of immune cells and synovial inflammation which leads to progressive disability. Current treatments improve the disease outcome, but the unmet medical need is still high. New discoveries over the last decade have revealed the major impact of cellular metabolism on immune cell functions. So far, a comprehensive understanding of metabolic changes during disease development, especially in the diseased microenvironment, is still limited. Therefore, we studied the longitudinal metabolic changes during the development of murine arthritis by integrating metabolomics and transcriptomics data. We identified an early change in macrophage pathways which was accompanied by oxidative stress, a drop in NAD+ level and induction of glucose transporters. We discovered inhibition of SIRT1, a NAD-dependent histone deacetylase and confirmed its dysregulation in human macrophages and synovial tissues of RA patients. Mining this database should enable the discovery of novel metabolic targets and therapy opportunities in RA.
© 2022. The Author(s).
Conflict of interest statement
All authors are current or former employees of Sanofi and may hold shares and/or stock options in the company.
Figures
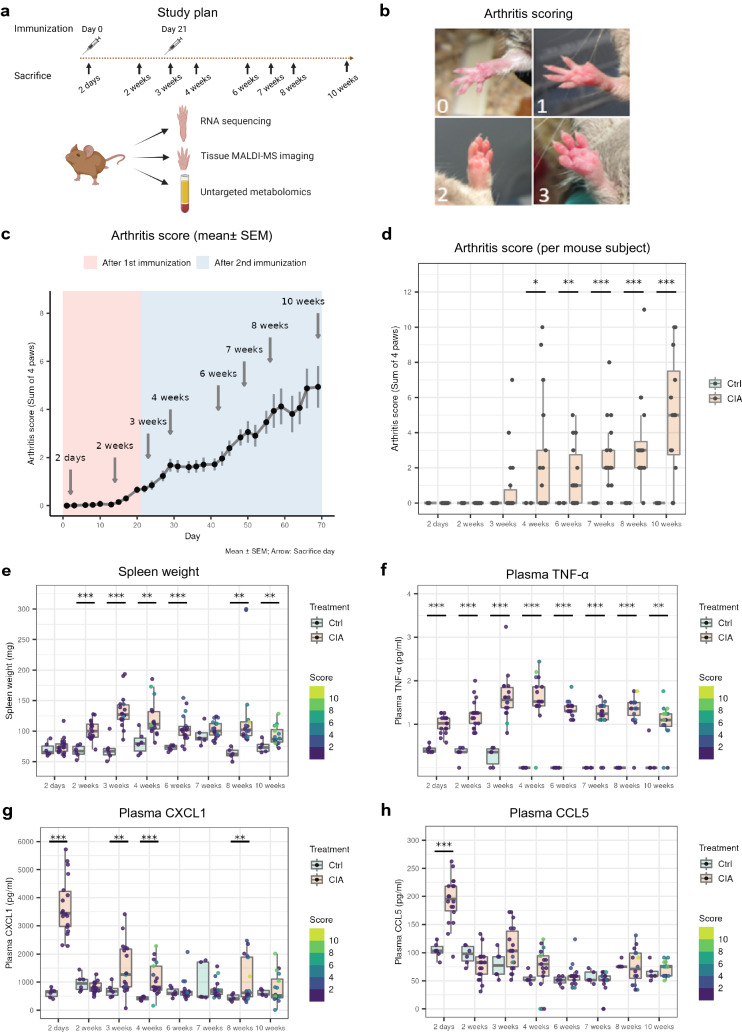

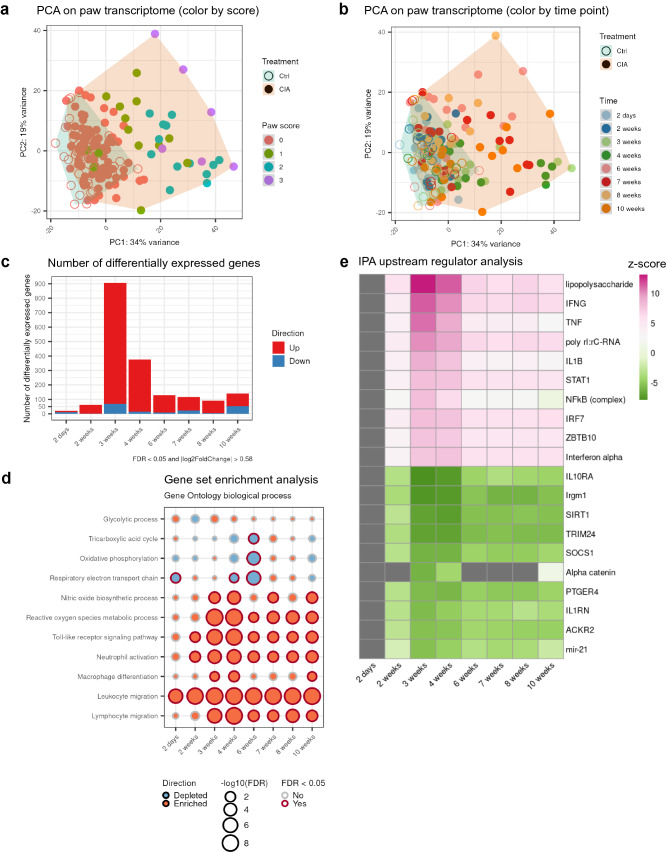
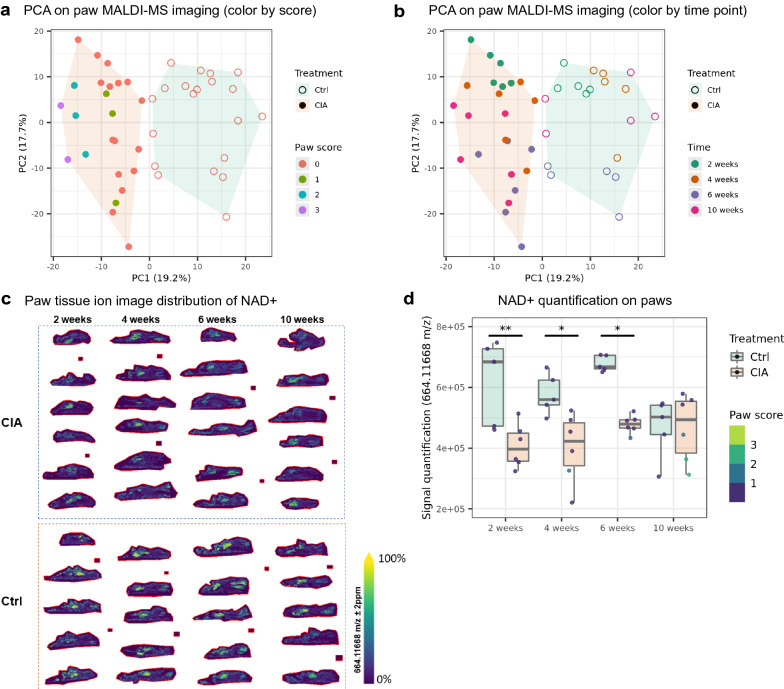
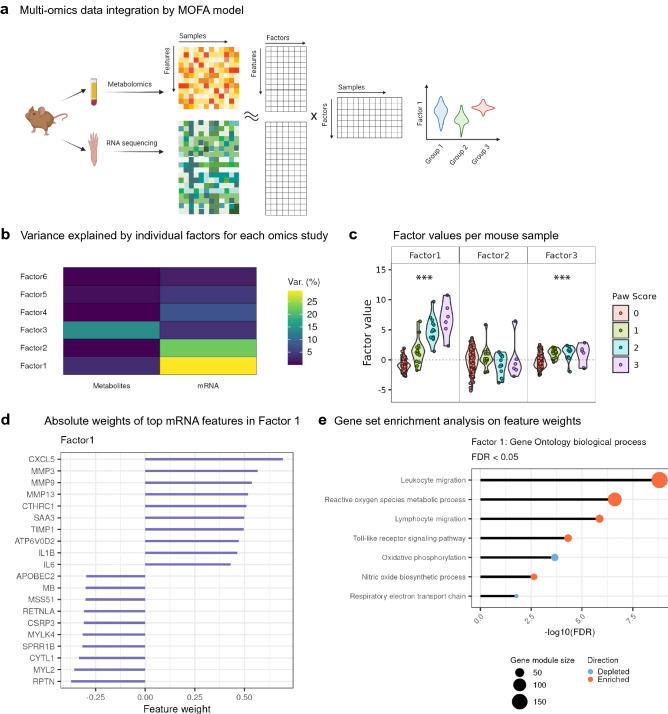


References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
