

Deciphering polymorphism in 61,157 Escherichia coli genomes via epistatic sequence landscapes
- PMID: 35821377
- PMCID: PMC9276797
- DOI: 10.1038/s41467-022-31643-3
Deciphering polymorphism in 61,157 Escherichia coli genomes via epistatic sequence landscapes
Abstract
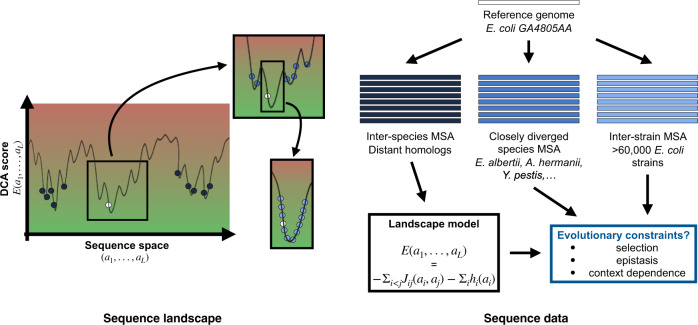
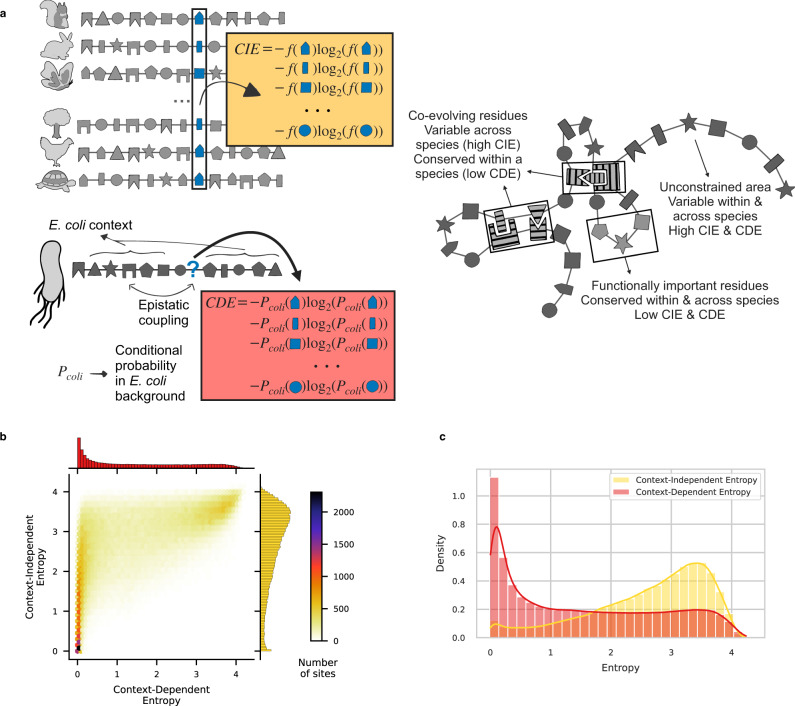
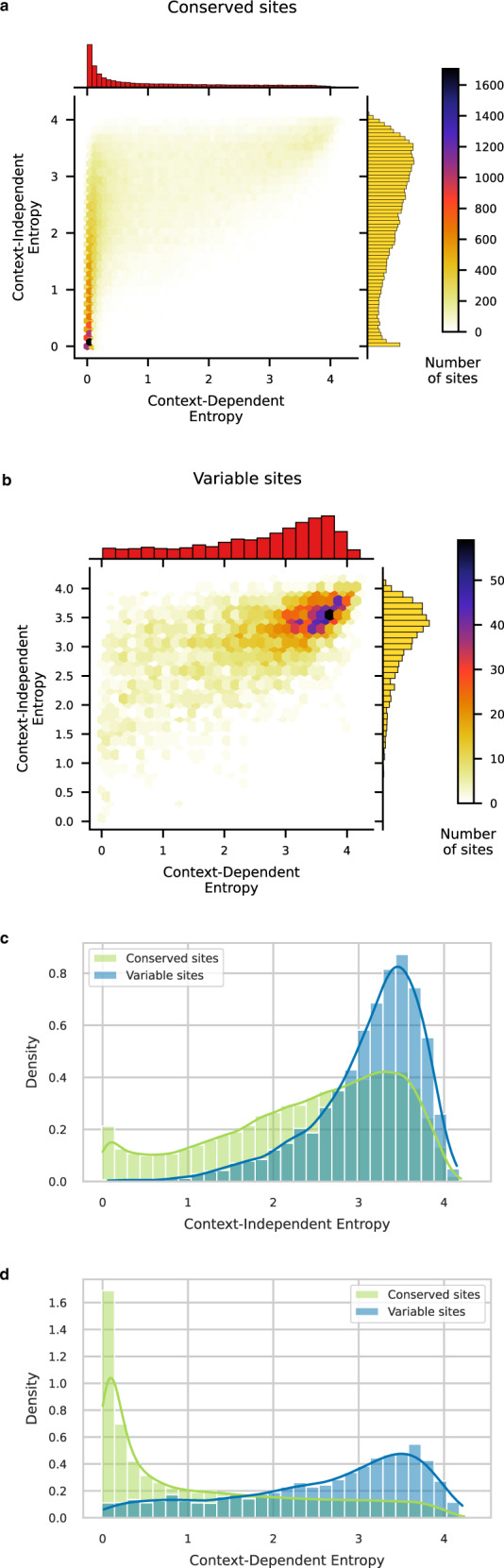
Characterizing the effect of mutations is key to understand the evolution of protein sequences and to separate neutral amino-acid changes from deleterious ones. Epistatic interactions between residues can lead to a context dependence of mutation effects. Context dependence constrains the amino-acid changes that can contribute to polymorphism in the short term, and the ones that can accumulate between species in the long term. We use computational approaches to accurately predict the polymorphisms segregating in a panel of 61,157 Escherichia coli genomes from the analysis of distant homologues. By comparing a context-aware Direct-Coupling Analysis modelling to a non-epistatic approach, we show that the genetic context strongly constrains the tolerable amino acids in 30% to 50% of amino-acid sites. The study of more distant species suggests the gradual build-up of genetic context over long evolutionary timescales by the accumulation of small epistatic contributions.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Mayr, E. How to carry out the adaptationist program? The American Naturalist121, 324–334 (1983).
-
- Kimura, M. The Neutral Theory of Molecular Evolution (Cambridge University Press, 1983).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
