

An atlas of endogenous DNA double-strand breaks arising during human neural cell fate determination
- PMID: 35821502
- PMCID: PMC9276747
- DOI: 10.1038/s41597-022-01508-x
An atlas of endogenous DNA double-strand breaks arising during human neural cell fate determination
Abstract
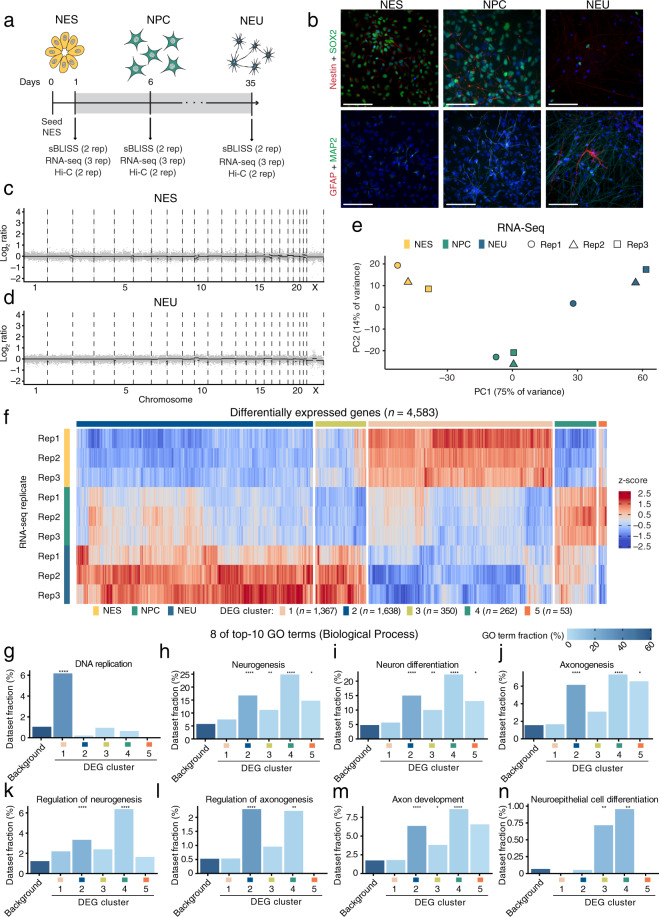
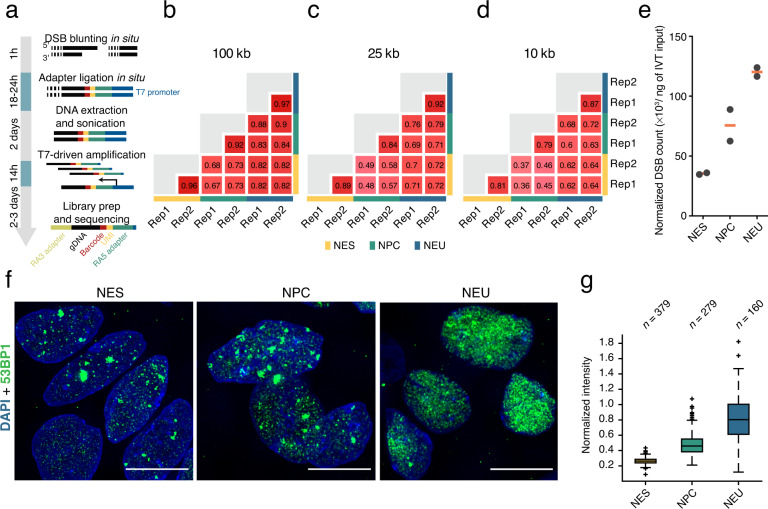
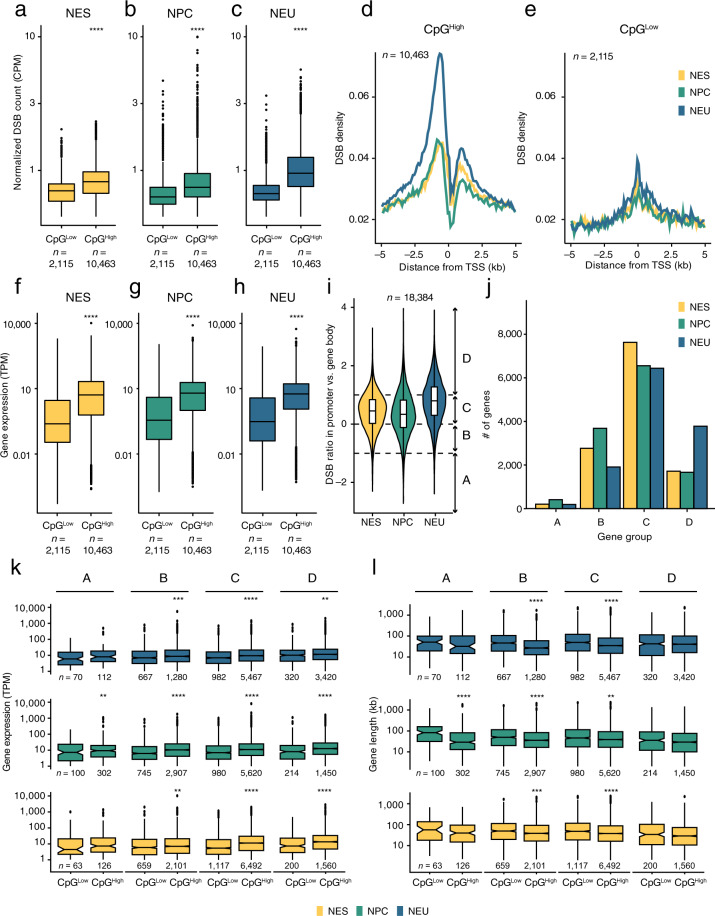
Endogenous DNA double-strand breaks (DSBs) occurring in neural cells have been implicated in the pathogenesis of neurodevelopmental disorders (NDDs). Currently, a genomic map of endogenous DSBs arising during human neurogenesis is missing. Here, we applied in-suspension Breaks Labeling In Situ and Sequencing (sBLISS), RNA-Seq, and Hi-C to chart the genomic landscape of DSBs and relate it to gene expression and genome architecture in 2D cultures of human neuroepithelial stem cells (NES), neural progenitor cells (NPC), and post-mitotic neural cells (NEU). Endogenous DSBs were enriched at the promoter and along the gene body of transcriptionally active genes, at the borders of topologically associating domains (TADs), and around chromatin loop anchors. NDD risk genes harbored significantly more DSBs in comparison to other protein-coding genes, especially in NEU cells. We provide sBLISS, RNA-Seq, and Hi-C datasets for each differentiation stage, and all the scripts needed to reproduce our analyses. Our datasets and tools represent a unique resource that can be harnessed to investigate the role of genome fragility in the pathogenesis of NDDs.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Gothe HJ, et al. Spatial Chromosome Folding and Active Transcription Drive DNA Fragility and Formation of Oncogenic MLL Translocations. Mol. Cell. 2019;75:267–283.e12. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
