

Implementation of Telescoping Boxes in Adaptive Steered Molecular Dynamics
- PMID: 35830368
- PMCID: PMC9369075
- DOI: 10.1021/acs.jctc.2c00498
Implementation of Telescoping Boxes in Adaptive Steered Molecular Dynamics
Abstract
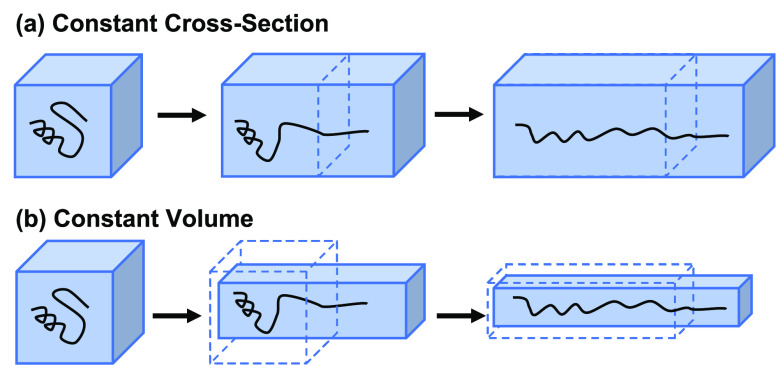
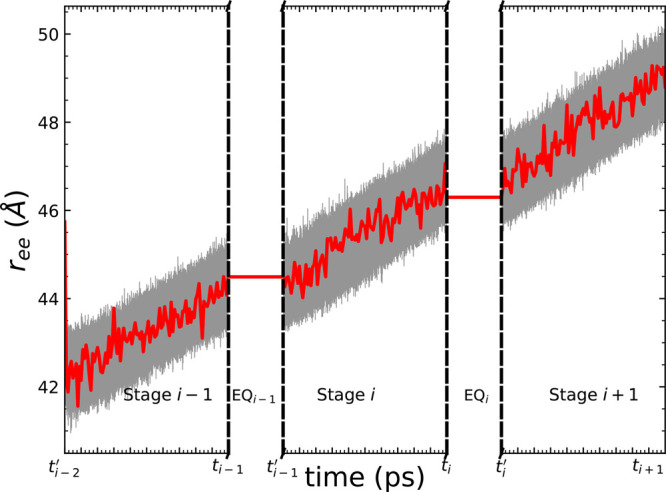
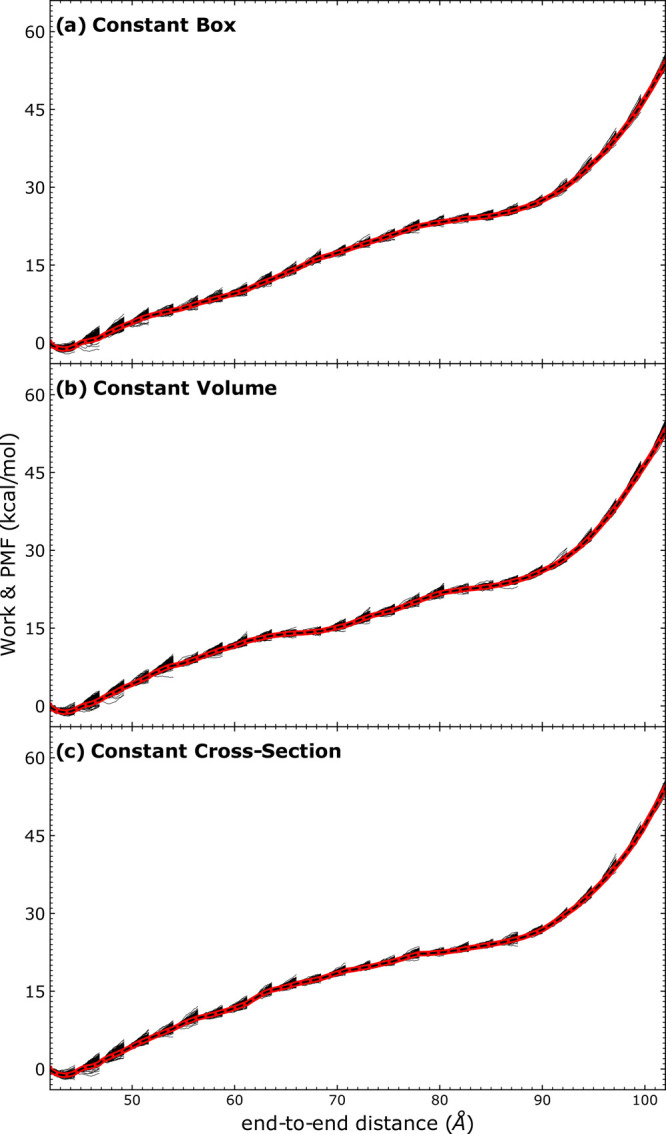
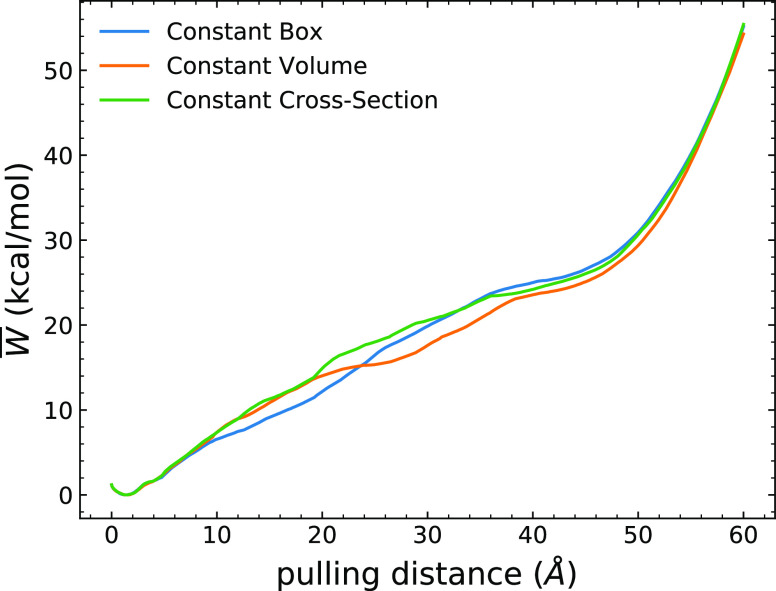
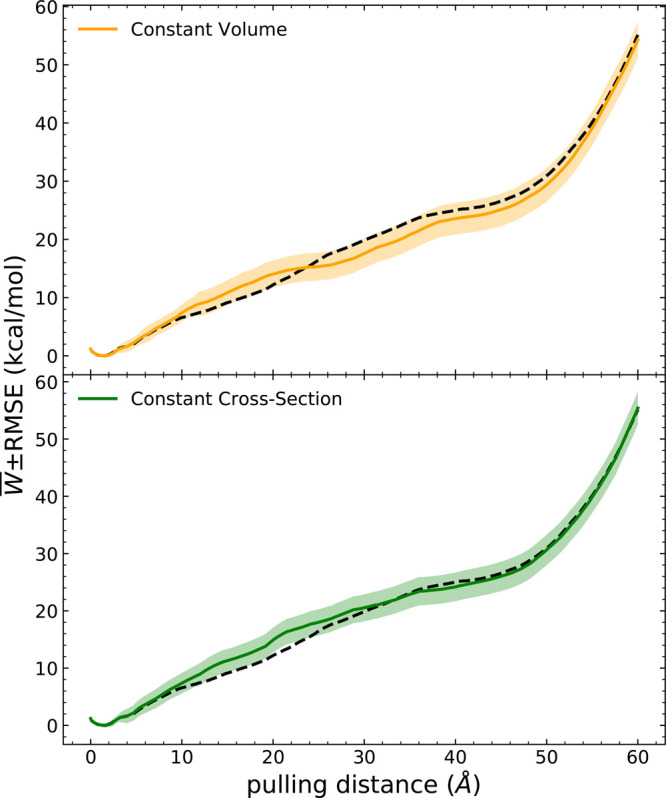
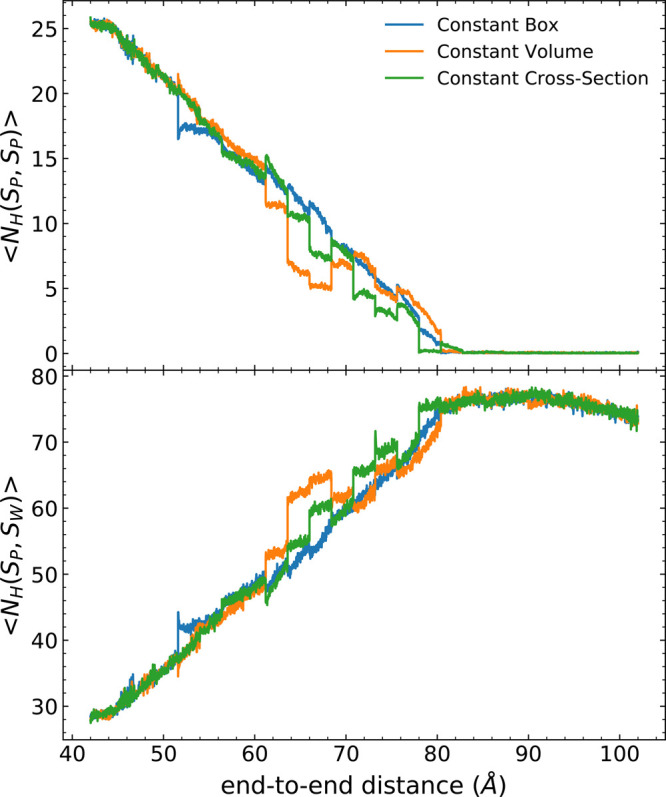
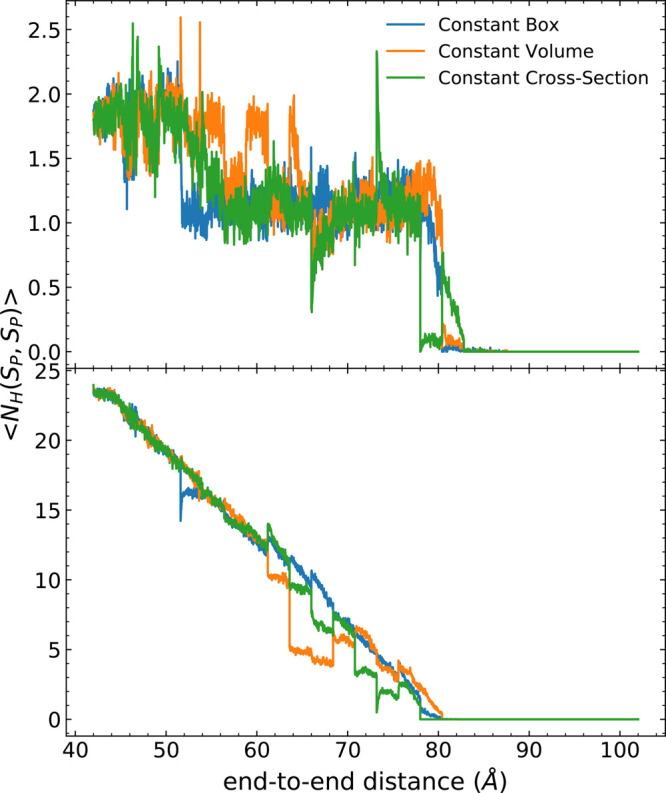
Long-time dynamical processes, such as those involving protein unfolding and ligand interactions, can be accelerated and realized through steered molecular dynamics (SMD). The challenge has been the extraction of information from such simulations that generalize for complex nonequilibrium processes. The use of Jarzynski's equality opened the possibility of determining the free energy along the steered coordinate, but sampling over the nonequilibrium trajectories is slow to converge. Adaptive steered molecular dynamics (ASMD) and other related techniques have been introduced to overcome this challenge through the use of stages. Here, we take advantage of these stages to address the numerical cost that arises from the required use of very large solvent boxes. We introduce telescoping box schemes within adaptive steered molecular dynamics (ASMD) in which we adjust the solvent box between stages and thereby vary (and optimize) the required number of solvent molecules. We have benchmarked the method on a relatively long α-helical peptide, Ala30, with respect to the potential of mean force and hydrogen bonds. We show that the use of telescoping boxes introduces little numerical error while significantly reducing the computational cost.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
 .
.
Similar articles
-
Constrained Unfolding of a Helical Peptide: Implicit versus Explicit Solvents.PLoS One. 2015 May 13;10(5):e0127034. doi: 10.1371/journal.pone.0127034. eCollection 2015. PLoS One. 2015. PMID: 25970521 Free PMC article.
-
Multiple branched adaptive steered molecular dynamics.J Chem Phys. 2014 Aug 14;141(6):064101. doi: 10.1063/1.4891807. J Chem Phys. 2014. PMID: 25134545
-
Benchmarking Adaptive Steered Molecular Dynamics (ASMD) on CHARMM Force Fields.Chemphyschem. 2022 Sep 5;23(17):e202200175. doi: 10.1002/cphc.202200175. Epub 2022 Jul 5. Chemphyschem. 2022. PMID: 35594194 Free PMC article.
-
Steered Molecular Dynamics Methods Applied to Enzyme Mechanism and Energetics.Methods Enzymol. 2016;578:123-43. doi: 10.1016/bs.mie.2016.05.029. Epub 2016 Jun 11. Methods Enzymol. 2016. PMID: 27497165 Review.
-
Taming Rugged Free Energy Landscapes Using an Average Force.Acc Chem Res. 2019 Nov 19;52(11):3254-3264. doi: 10.1021/acs.accounts.9b00473. Epub 2019 Nov 4. Acc Chem Res. 2019. PMID: 31680510 Review.
Cited by
-
Shocker─A Molecular Dynamics Protocol and Tool for Accelerating and Analyzing the Effects of Osmotic Shocks.J Chem Theory Comput. 2024 Jan 9;20(1):212-223. doi: 10.1021/acs.jctc.3c00961. Epub 2023 Dec 18. J Chem Theory Comput. 2024. PMID: 38109481 Free PMC article.
-
Molecular Modelling of Ionic Liquids: Situations When Charge Scaling Seems Insufficient.Molecules. 2023 Jan 13;28(2):800. doi: 10.3390/molecules28020800. Molecules. 2023. PMID: 36677859 Free PMC article.
-
Tertiary Plasticity Drives the Efficiency of Enterocin 7B Interactions with Lipid Membranes.J Phys Chem B. 2024 Mar 7;128(9):2100-2113. doi: 10.1021/acs.jpcb.3c08199. Epub 2024 Feb 27. J Phys Chem B. 2024. PMID: 38412510 Free PMC article.
-
Correlation between chemical denaturation and the unfolding energetics of Acanthamoeba actophorin.Biophys J. 2023 Jul 25;122(14):2921-2937. doi: 10.1016/j.bpj.2022.11.2941. Epub 2022 Dec 2. Biophys J. 2023. PMID: 36461639 Free PMC article.
-
Free energy and kinetic rate calculation via non-equilibrium molecular simulation: application to biomolecules.Biophys Rev. 2022 Dec 29;14(6):1303-1314. doi: 10.1007/s12551-022-01036-3. eCollection 2022 Dec. Biophys Rev. 2022. PMID: 36659997 Free PMC article. Review.
References
-
- Frenkel D. Simulations: The Dark Side. Eur. Phys. J. Plus 2013, 128, 1–10. 10.1140/epjp/i2013-13010-8. - DOI
-
- Bernardi R. C.; Durner E.; Schoeler C.; Malinowska K. H.; Carvalho B. G.; Bayer E. A.; Luthey-Schulten Z.; Gaub H. E.; Nash M. A. Mechanisms of Nanonewton Mechanostability in a Protein Complex Revealed by Molecular Dynamics Simulations and Single-Molecule Force Spectroscopy. J. Am. Chem. Soc. 2019, 141, 14752–14763. 10.1021/jacs.9b06776. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
