

Mislocalization of protein kinase A drives pathology in Cushing's syndrome
- PMID: 35830806
- PMCID: PMC9311266
- DOI: 10.1016/j.celrep.2022.111073
Mislocalization of protein kinase A drives pathology in Cushing's syndrome
Abstract
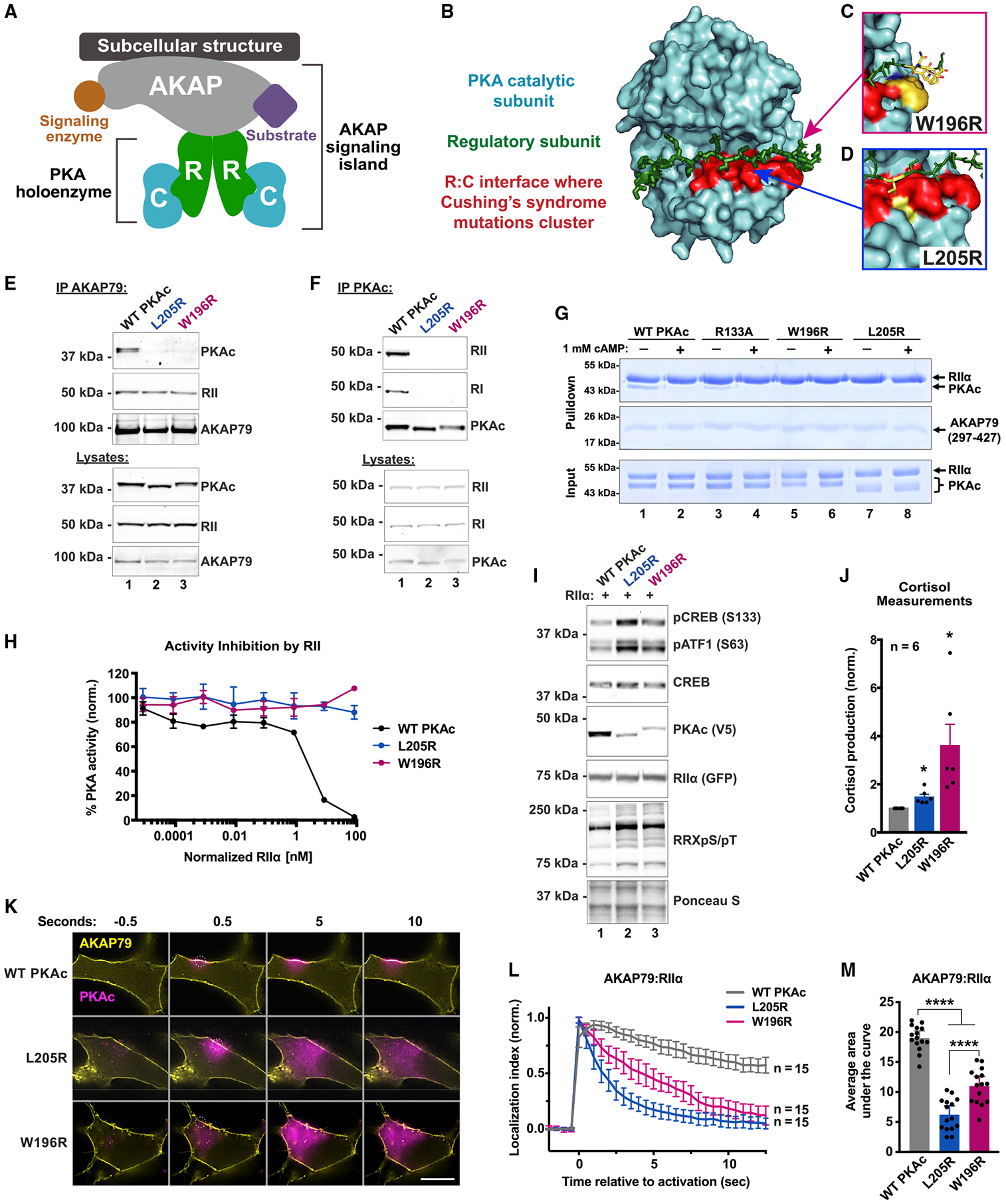
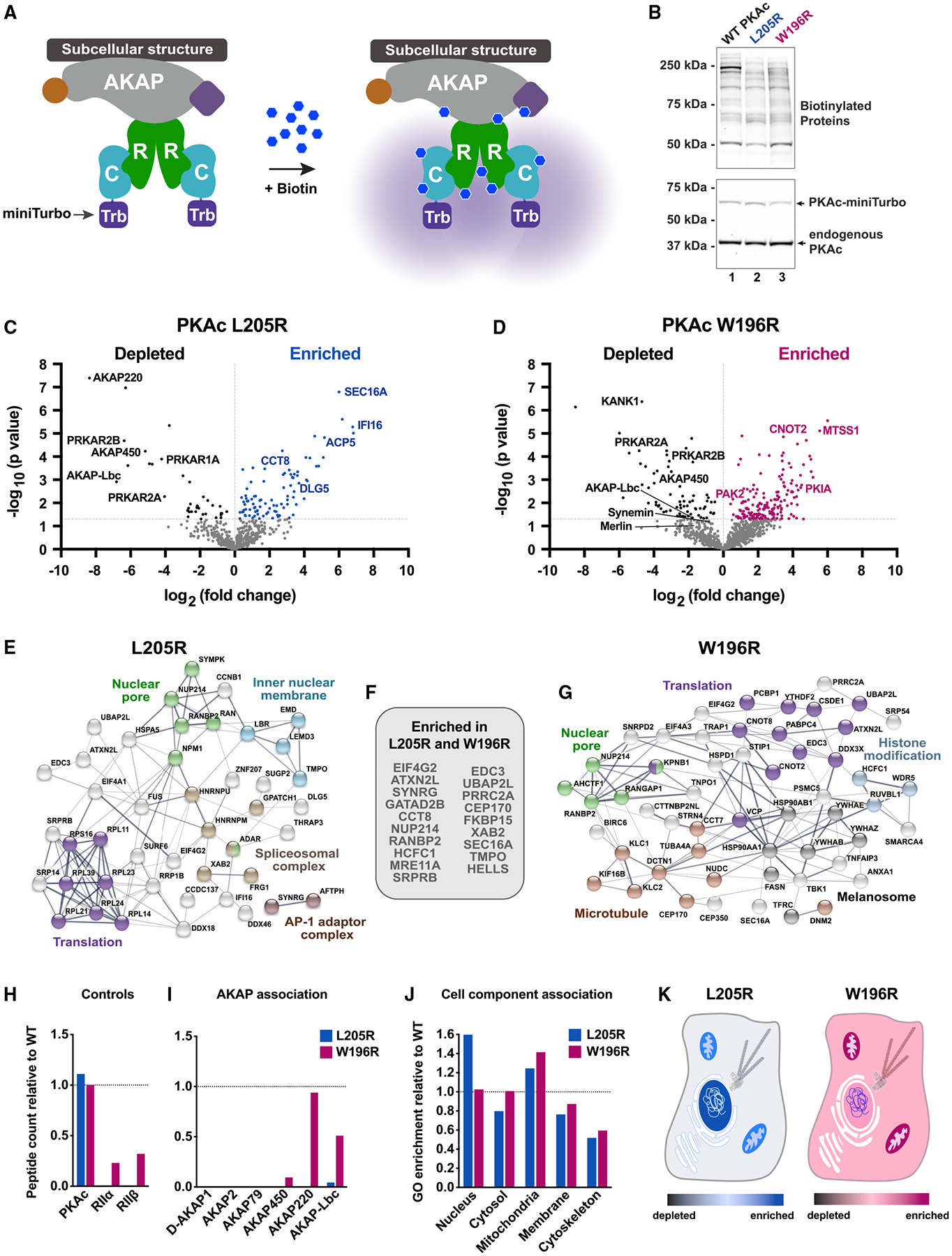
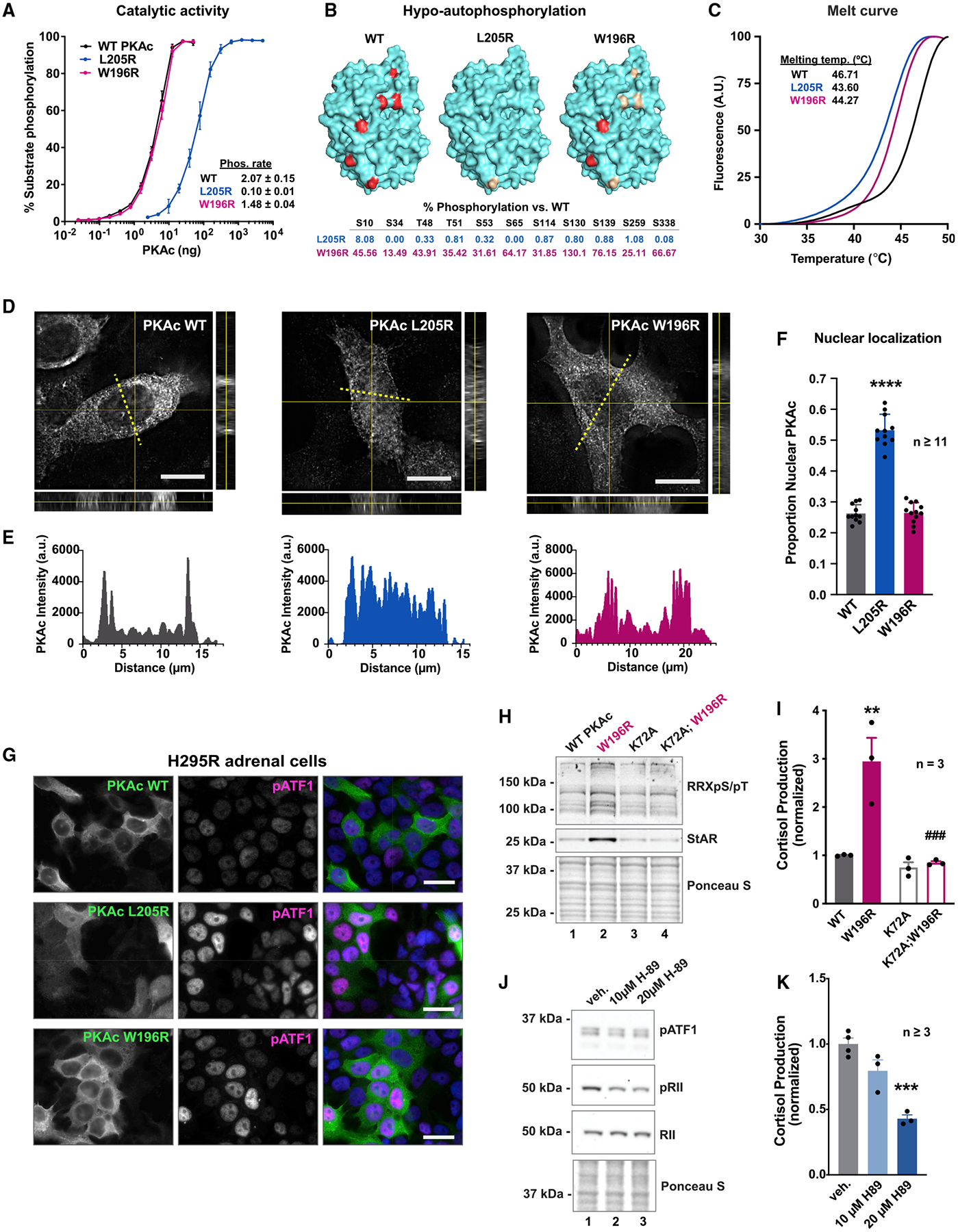
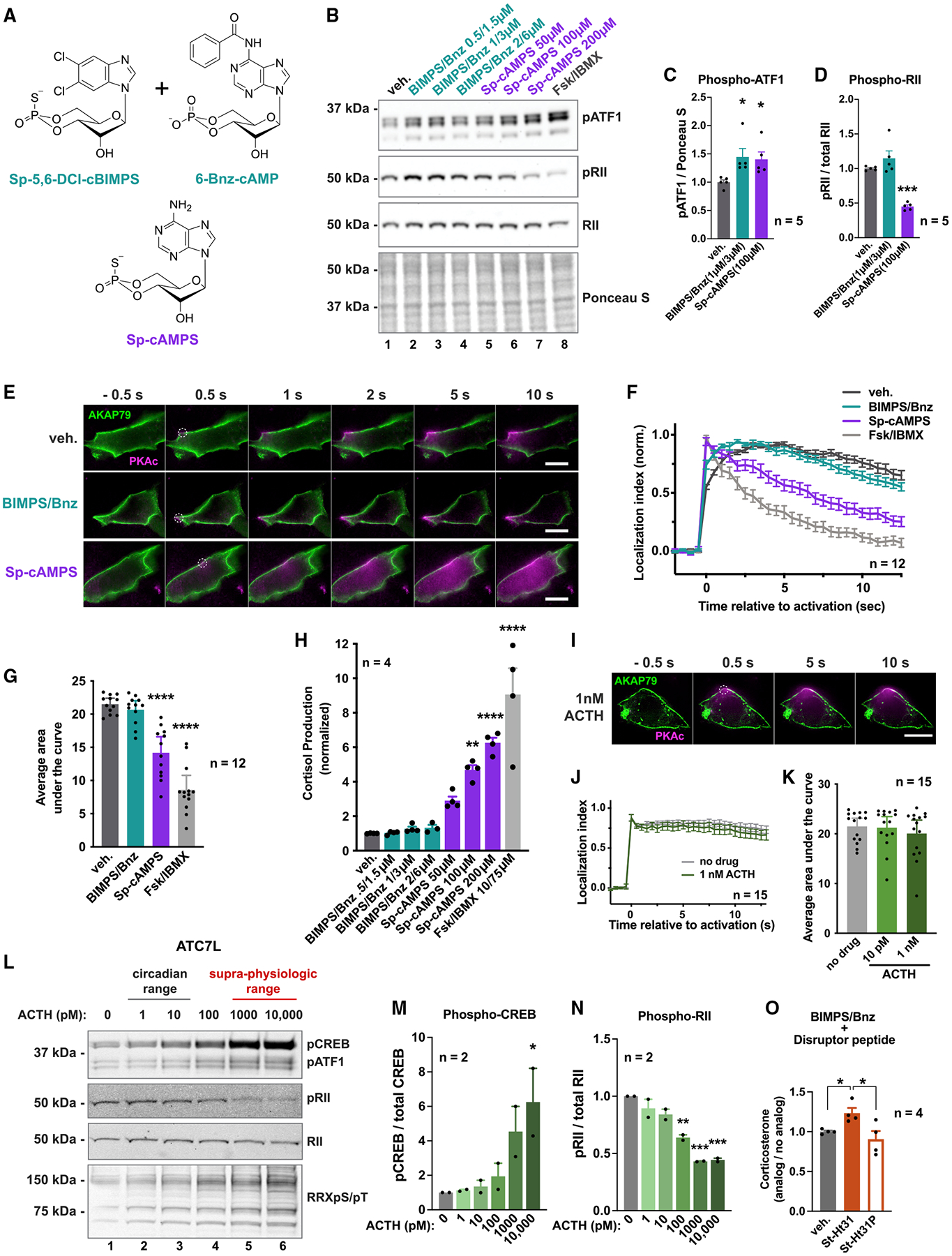
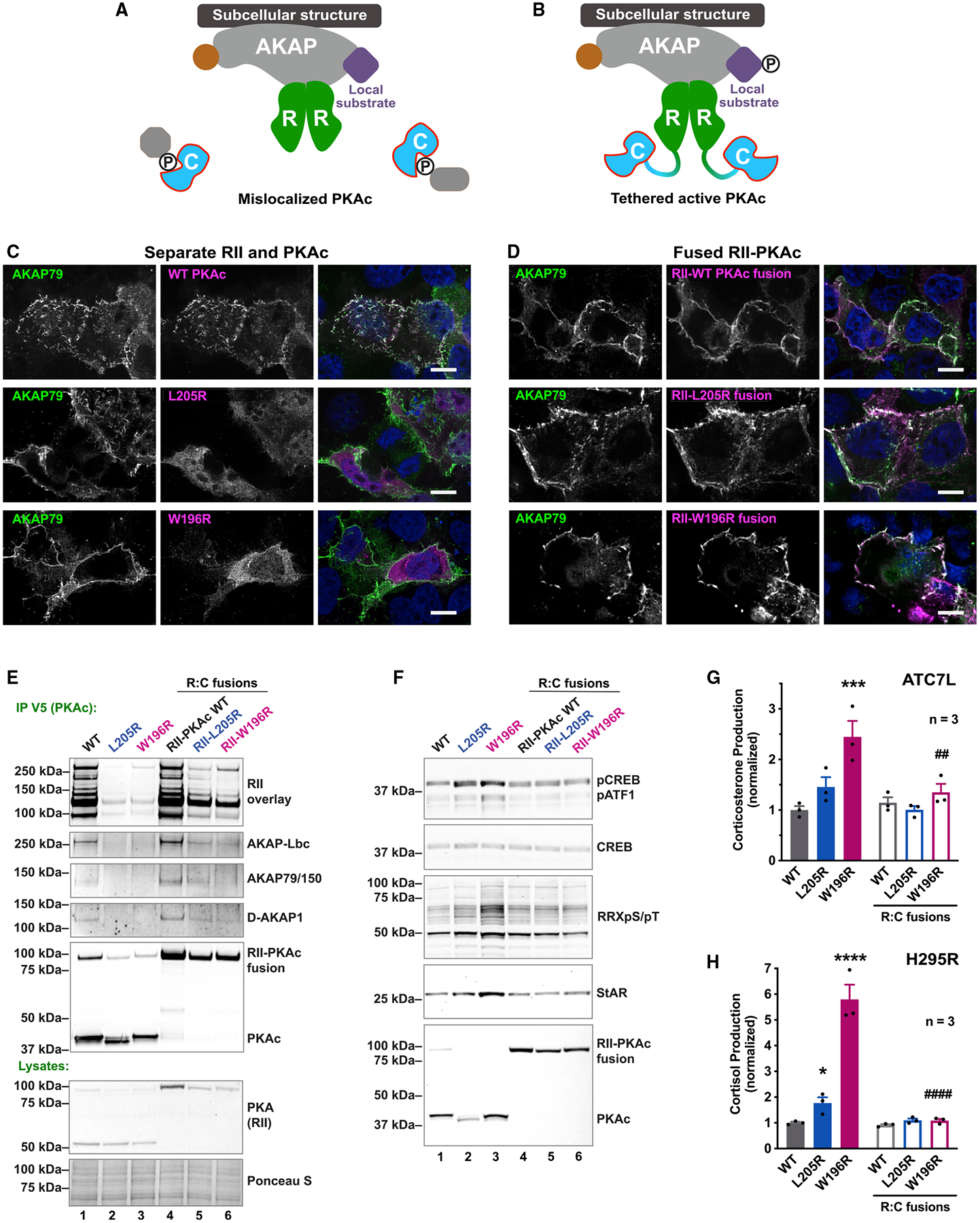
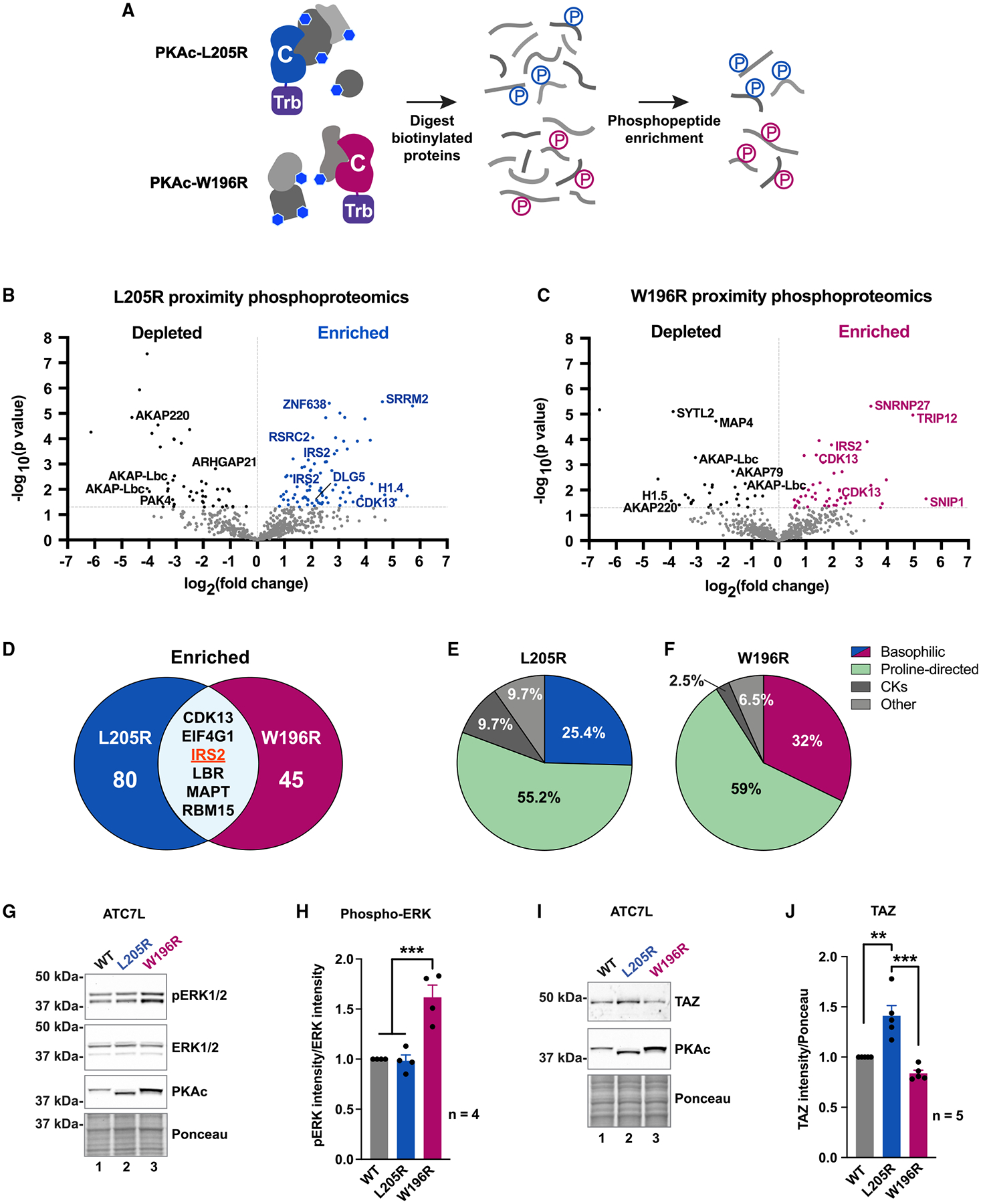
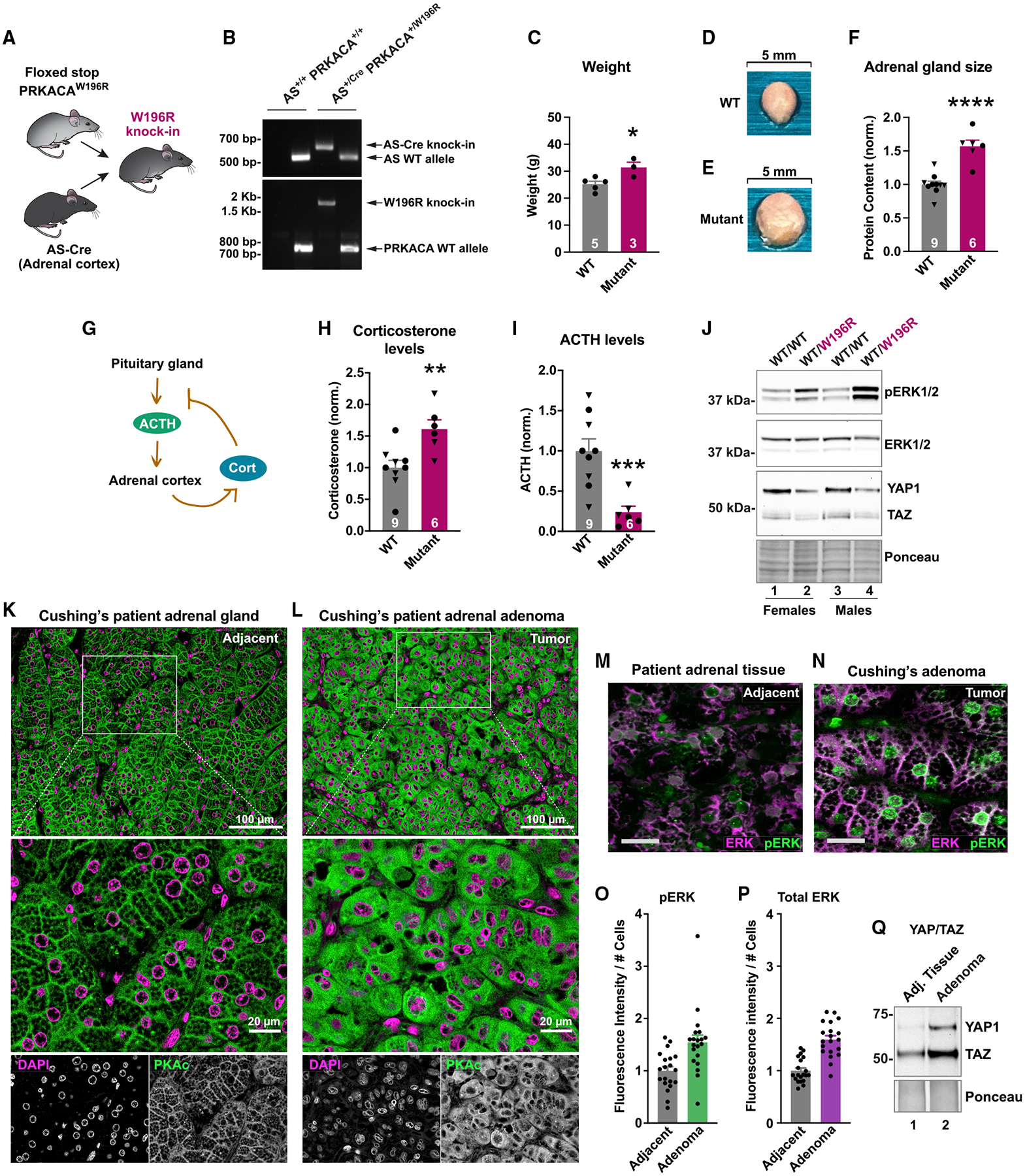
Mutations in the catalytic subunit of protein kinase A (PKAc) drive the stress hormone disorder adrenal Cushing's syndrome. We define mechanisms of action for the PKAc-L205R and W196R variants. Proximity proteomic techniques demonstrate that both Cushing's mutants are excluded from A kinase-anchoring protein (AKAP)-signaling islands, whereas live-cell photoactivation microscopy reveals that these kinase mutants indiscriminately diffuse throughout the cell. Only cAMP analog drugs that displace native PKAc from AKAPs enhance cortisol release. Rescue experiments that incorporate PKAc mutants into AKAP complexes abolish cortisol overproduction, indicating that kinase anchoring restores normal endocrine function. Analyses of adrenal-specific PKAc-W196R knockin mice and Cushing's syndrome patient tissue reveal defective signaling mechanisms of the disease. Surprisingly each Cushing's mutant engages a different mitogenic-signaling pathway, with upregulation of YAP/TAZ by PKAc-L205R and ERK kinase activation by PKAc-W196R. Thus, aberrant spatiotemporal regulation of each Cushing's variant promotes the transmission of distinct downstream pathogenic signals.
Keywords: CP: Cell biology; CP: Metabolism; PRKACA; adrenal; anchoring; cortisol; photoactivation; proteomics; proximity biotinylation; scaffold; signaling; stress hormone.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DMF, Dessauer CW, and Scott JD (2006). Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Molecular cell 23, 925–931. 10.1016/j.molcel.2006.07.025. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- BB/S018514/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- R01 DK119192/DK/NIDDK NIH HHS/United States
- S10 OD021502/OD/NIH HHS/United States
- F32 DK121415/DK/NIDDK NIH HHS/United States
- T32 DK007247/DK/NIDDK NIH HHS/United States
- C1443/A22095/CRUK_/Cancer Research UK/United Kingdom
- R01 DK119186/DK/NIDDK NIH HHS/United States
- BB/T018127/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- R01 GM129090/GM/NIGMS NIH HHS/United States
- T32 GM007270/GM/NIGMS NIH HHS/United States
- R01 DK123694/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
