

Molecular and clinical profiling in a large cohort of Asian Indians with glycogen storage disorders
- PMID: 35834487
- PMCID: PMC9282608
- DOI: 10.1371/journal.pone.0270373
Molecular and clinical profiling in a large cohort of Asian Indians with glycogen storage disorders
Abstract
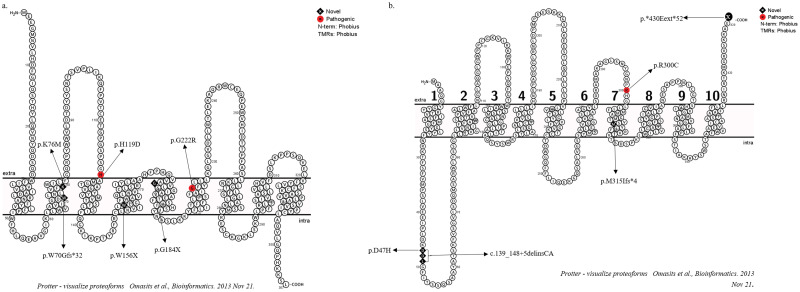
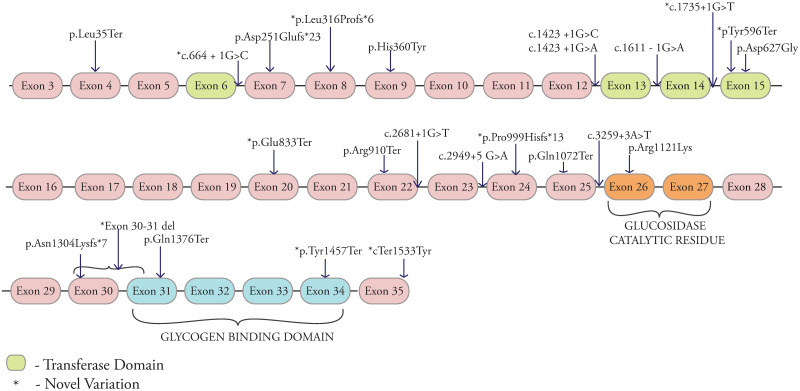
Glycogen storage disorders occur due to enzyme deficiencies in the glycogenolysis and gluconeogenesis pathway, encoded by 26 genes. GSD's present with overlapping phenotypes with variable severity. In this series, 57 individuals were molecularly confirmed for 7 GSD subtypes and their demographic data, clinical profiles and genotype-phenotype co-relations are studied. Genomic DNA from venous blood samples was isolated from clinically affected individuals. Targeted gene panel sequencing covering 23 genes and Sanger sequencing were employed. Various bioinformatic tools were used to predict pathogenicity for new variations. Close parental consanguinity was seen in 76%. Forty-nine pathogenic variations were detected of which 27 were novel. Variations were spread across GSDIa, Ib, III, VI, IXa, b and c. The largest subgroup was GSDIII in 28 individuals with 24 variations (12 novel) in AGL. The 1620+1G>C intronic variation was observed in 5 with GSDVI (PYGL). A total of eleven GSDIX are described with the first Indian report of type IXb. This is the largest study of GSDs from India. High levels of consanguinity in the local population and employment of targeted sequencing panels accounted for the range of GSDs reported here.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Smit GPA, Rake JP, Akman HO, DiMauro S. The glycogen storage diseases and related disorders. Fernandes J, Saudubray J-M, van den Berghe G, Walter JH (eds) Inborn metabolic diseases: diagnosis and treatment. Springer Medizin; Verlag, Heidelberg; 2006:103–116.
-
- Karthi Sellamuthu, Manimaran Paramasivam, Varalakshmi Perumal, Ganesh Ramaswamy, Kapoor Seema, Goyal Manisha, et al.. Mutational spectrum and identification of five novel mutations in G6PC1 gene from a cohort of Glycogen Storage Disease Type 1a. Gene. 2019; 700:7–16; doi: 10.1016/j.gene.2019.03.029 - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
