

Auxotrophic and prototrophic conditional genetic networks reveal the rewiring of transcription factors in Escherichia coli
- PMID: 35835781
- PMCID: PMC9283627
- DOI: 10.1038/s41467-022-31819-x
Auxotrophic and prototrophic conditional genetic networks reveal the rewiring of transcription factors in Escherichia coli
Abstract
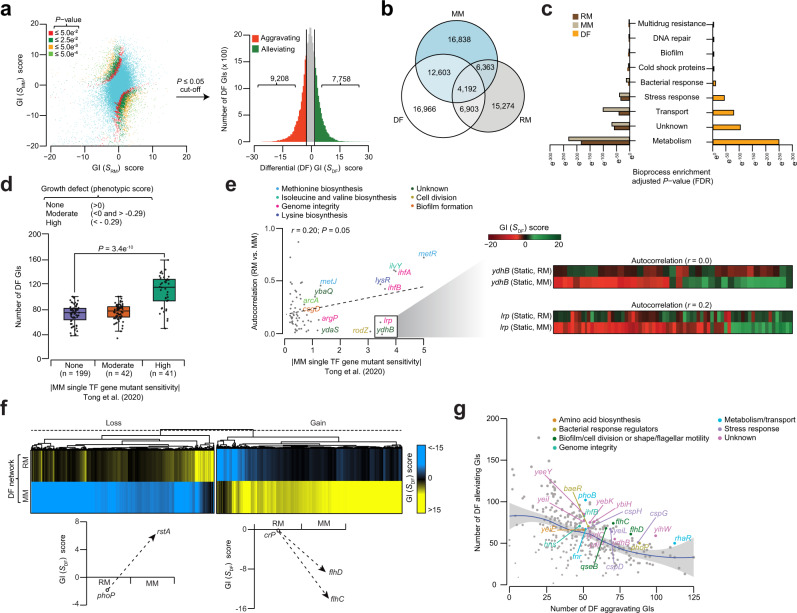
Bacterial transcription factors (TFs) are widely studied in Escherichia coli. Yet it remains unclear how individual genes in the underlying pathways of TF machinery operate together during environmental challenge. Here, we address this by applying an unbiased, quantitative synthetic genetic interaction (GI) approach to measure pairwise GIs among all TF genes in E. coli under auxotrophic (rich medium) and prototrophic (minimal medium) static growth conditions. The resulting static and differential GI networks reveal condition-dependent GIs, widespread changes among TF genes in metabolism, and new roles for uncharacterized TFs (yjdC, yneJ, ydiP) as regulators of cell division, putrescine utilization pathway, and cold shock adaptation. Pan-bacterial conservation suggests TF genes with GIs are co-conserved in evolution. Together, our results illuminate the global organization of E. coli TFs, and remodeling of genetic backup systems for TFs under environmental change, which is essential for controlling the bacterial transcriptional regulatory circuits.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests
Figures





Similar articles
-
Conditional Epistatic Interaction Maps Reveal Global Functional Rewiring of Genome Integrity Pathways in Escherichia coli.Cell Rep. 2016 Jan 26;14(3):648-661. doi: 10.1016/j.celrep.2015.12.060. Epub 2016 Jan 8. Cell Rep. 2016. PMID: 26774489
-
Systematic discovery of uncharacterized transcription factors in Escherichia coli K-12 MG1655.Nucleic Acids Res. 2018 Nov 16;46(20):10682-10696. doi: 10.1093/nar/gky752. Nucleic Acids Res. 2018. PMID: 30137486 Free PMC article.
-
Quantifying the regulatory role of individual transcription factors in Escherichia coli.Cell Rep. 2021 Nov 9;37(6):109952. doi: 10.1016/j.celrep.2021.109952. Cell Rep. 2021. PMID: 34758318 Free PMC article.
-
Hierarchy of transcription factor network in Escherichia coli K-12: H-NS-mediated silencing and Anti-silencing by global regulators.FEMS Microbiol Rev. 2021 Nov 23;45(6):fuab032. doi: 10.1093/femsre/fuab032. FEMS Microbiol Rev. 2021. PMID: 34196371 Review.
-
Building a complete image of genome regulation in the model organism Escherichia coli.J Gen Appl Microbiol. 2018 Jan 15;63(6):311-324. doi: 10.2323/jgam.2017.01.002. Epub 2017 Sep 12. J Gen Appl Microbiol. 2018. PMID: 28904250 Review.
Cited by
-
Overview of the Molecular Mechanism of Bacterial Environmental Adaptation by Comprehensive Analysis.Int J Mol Sci. 2023 Apr 20;24(8):7602. doi: 10.3390/ijms24087602. Int J Mol Sci. 2023. PMID: 37108762 Free PMC article.
-
A Cryptic Prophage Transcription Factor Drives Phenotypic Changes via Host Gene Regulation.bioRxiv [Preprint]. 2024 Sep 21:2024.09.21.614188. doi: 10.1101/2024.09.21.614188. bioRxiv. 2024. PMID: 39345586 Free PMC article. Preprint.
-
Antipsychotic quetiapine alters the mouse fecal resistome by impacting antibiotic efflux, cell membrane, and cell wall synthesis genes.Microbiol Spectr. 2024 Jan 11;12(1):e0380423. doi: 10.1128/spectrum.03804-23. Epub 2023 Dec 15. Microbiol Spectr. 2024. PMID: 38099619 Free PMC article.
-
In vitro transcription-based biosensing of glycolate for prototyping of a complex enzyme cascade.Synth Biol (Oxf). 2024 Sep 20;9(1):ysae013. doi: 10.1093/synbio/ysae013. eCollection 2024. Synth Biol (Oxf). 2024. PMID: 39399720 Free PMC article.
-
Transcriptional Regulation in Bacteria.Microorganisms. 2024 Dec 6;12(12):2514. doi: 10.3390/microorganisms12122514. Microorganisms. 2024. PMID: 39770717 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
