

Mechanisms of Ferroptosis and Emerging Links to the Pathology of Neurodegenerative Diseases
- PMID: 35837484
- PMCID: PMC9273851
- DOI: 10.3389/fnagi.2022.904152
Mechanisms of Ferroptosis and Emerging Links to the Pathology of Neurodegenerative Diseases
Abstract
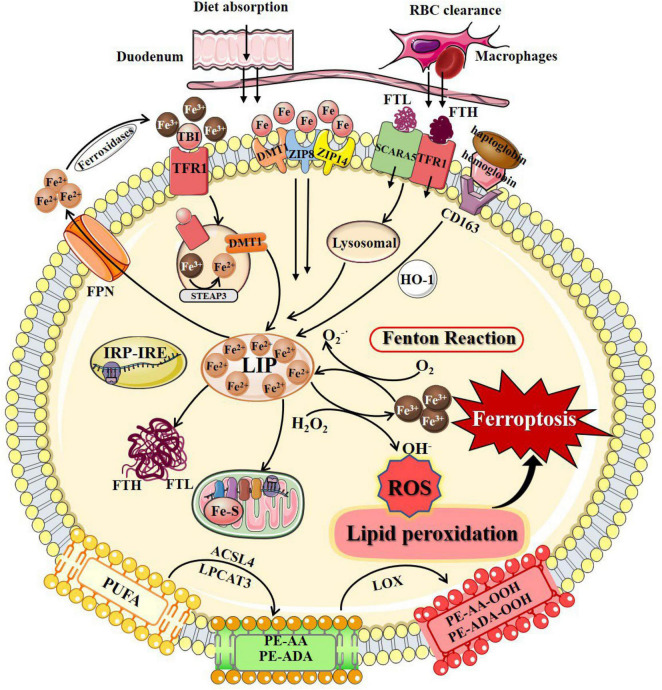
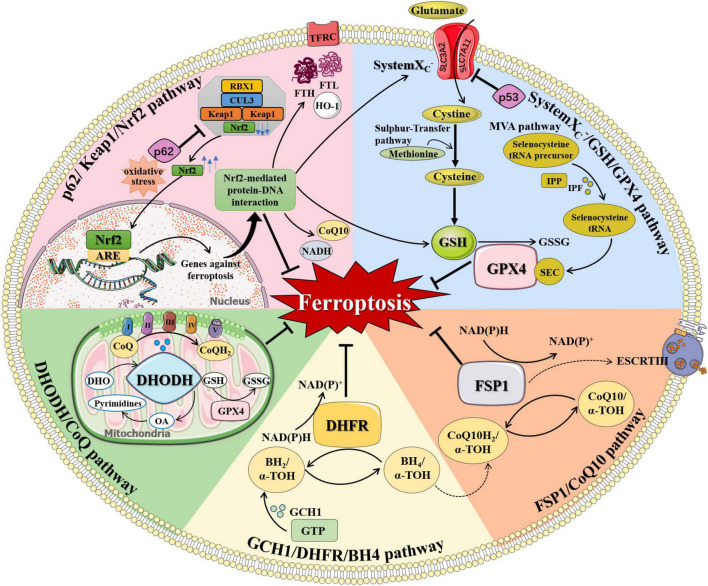
Neurodegenerative diseases are a diverse class of diseases attributed to chronic progressive neuronal degeneration and synaptic loss in the brain and/or spinal cord, including Alzheimer's disease, Parkinson's disease, Huntington's disease, amyotrophic lateral sclerosis and multiple sclerosis. The pathogenesis of neurodegenerative diseases is complex and diverse, often involving mitochondrial dysfunction, neuroinflammation, and epigenetic changes. However, the pathogenesis of neurodegenerative diseases has not been fully elucidated. Recently, accumulating evidence revealed that ferroptosis, a newly discovered iron-dependent and lipid peroxidation-driven type of programmed cell death, provides another explanation for the occurrence and progression of neurodegenerative diseases. Here, we provide an overview of the process and regulation mechanisms of ferroptosis, and summarize current research progresses that support the contribution of ferroptosis to the pathogenesis of neurodegenerative diseases. A comprehensive understanding of the emerging roles of ferroptosis in neurodegenerative diseases will shed light on the development of novel therapeutic technologies and strategies for slowing down the progression of these diseases.
Keywords: ferroptosis; iron metabolism; neurodegenerative diseases; oxidative stress; redox regulation.
Copyright © 2022 Sun, Xia, Basnet, Zheng, Huang and Liu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources