

Pan-cancer proteomic map of 949 human cell lines
- PMID: 35839778
- PMCID: PMC9387775
- DOI: 10.1016/j.ccell.2022.06.010
Pan-cancer proteomic map of 949 human cell lines
Abstract
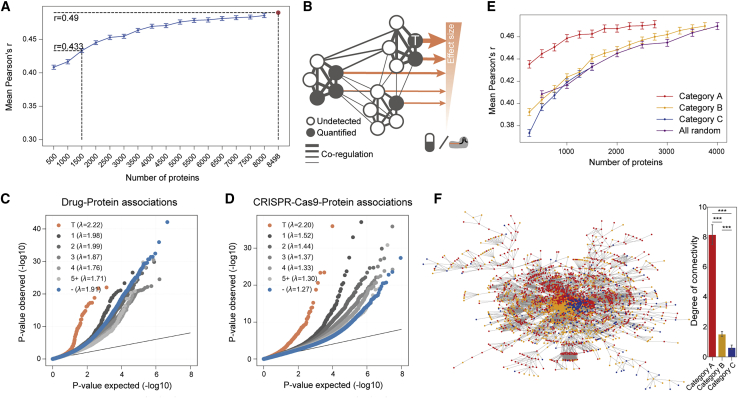
The proteome provides unique insights into disease biology beyond the genome and transcriptome. A lack of large proteomic datasets has restricted the identification of new cancer biomarkers. Here, proteomes of 949 cancer cell lines across 28 tissue types are analyzed by mass spectrometry. Deploying a workflow to quantify 8,498 proteins, these data capture evidence of cell-type and post-transcriptional modifications. Integrating multi-omics, drug response, and CRISPR-Cas9 gene essentiality screens with a deep learning-based pipeline reveals thousands of protein biomarkers of cancer vulnerabilities that are not significant at the transcript level. The power of the proteome to predict drug response is very similar to that of the transcriptome. Further, random downsampling to only 1,500 proteins has limited impact on predictive power, consistent with protein networks being highly connected and co-regulated. This pan-cancer proteomic map (ProCan-DepMapSanger) is a comprehensive resource available at https://cellmodelpassports.sanger.ac.uk.
Keywords: CRISPR-Cas9; cancer; cancer vulnerability; cell line; drug response; gene essentiality; mass spectrometry; proteomics.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.J.G. has received research funding from AstraZeneca, GSK, Astex Therapeutics, and Open Targets, a public-private initiative involving academia and industry, and is a co-founder of Mosaic Therapeutics. All other authors declare no competing interests.
Figures







References
-
- Aulchenko Y.S., Ripke S., Isaacs A., van Duijn C.M. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–1296. - PubMed
-
- Behan F.M., Iorio F., Picco G., Gonçalves E., Beaver C.M., Migliardi G., Santos R., Rao Y., Sassi F., Pinnelli M., et al. Prioritization of cancer therapeutic targets using CRISPR-cas9 screens. Nature. 2019;568:511–516. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
