

A biallelic loss-of-function variant in MYZAP is associated with a recessive form of severe dilated cardiomyopathy
- PMID: 35840178
- PMCID: PMC9528970
- DOI: 10.1101/mcs.a006221
A biallelic loss-of-function variant in MYZAP is associated with a recessive form of severe dilated cardiomyopathy
Abstract
Purpose: Dilated cardiomyopathy (DCM) is a primary disorder of the cardiac muscle, characterised by dilatation of the left ventricle and contractile dysfunction. About 50% of DCM cases can be attributed to monogenic causes, whereas the aetiology in the remaining patients remains unexplained.
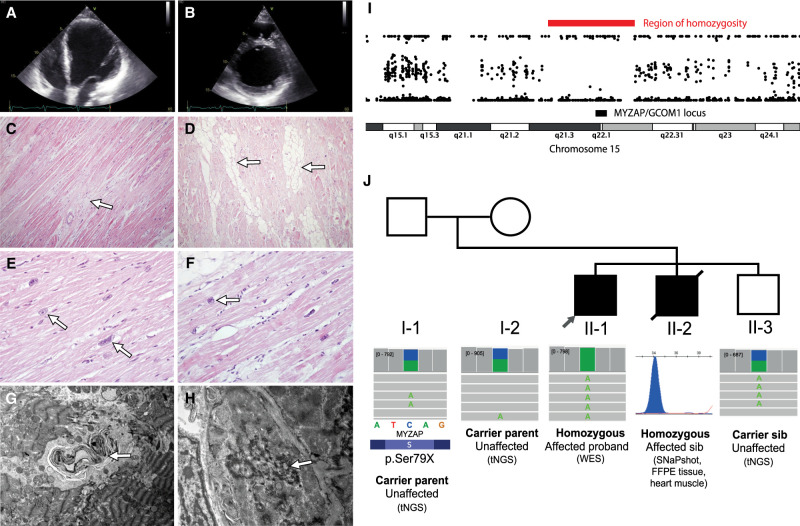
Methods: We report a family with two brothers affected by severe DCM with onset in the adolescent period. Using exome sequencing, we identified a homozygous premature termination variant in the MYZAP gene in both affected sibs. MYZAP encodes for myocardial zonula adherens protein - a conserved cardiac protein in the intercalated disc structure of cardiomyocytes.
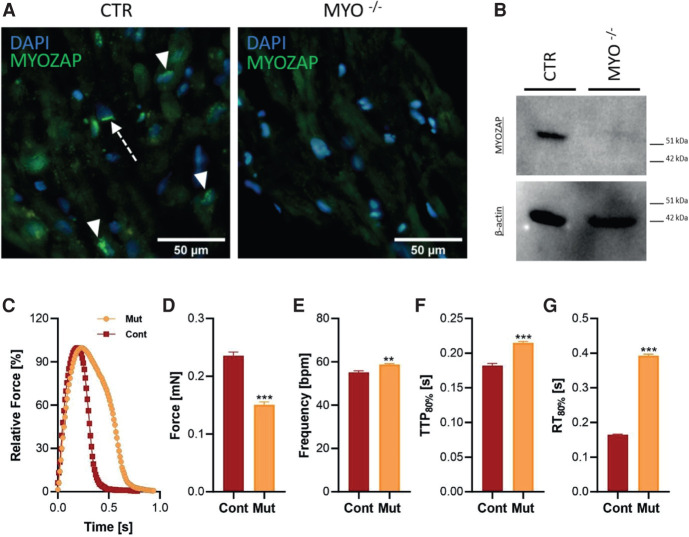
Results: The effect of the variant was demonstrated by light and electron microscopy of the heart muscle and immunohistochemical and Western blot analysis of MYZAP protein in the heart tissue of the proband. Functional characterization using patient-derived induced pluripotent stem cell cardiomyocytes revealed significantly lower force and longer time to peak contraction and relaxation consistent with severe contractile dysfunction.
Conclusion: We provide independent support for the role of biallelic loss-of-function MYZAP variants in dilated cardiomyopathy. This report extends the spectrum of cardiac disease associated with dysfunction of cardiac intercalated disc junction and sheds light on the mechanisms leading to DCM.
Keywords: Dilated cardiomyopathy.
Cold Spring Harbor Laboratory Press.
Figures
References
-
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kühl U, Maisch B, McKenna WJ, et al. 2008. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 29: 270–276. 10.1093/eurheartj/ehm342 - DOI - PubMed
-
- Heliö K, Mäyränpää MI, Saarinen I, Ahonen S, Junnila H, Tommiska J, Weckström S, Holmström M, Toivonen M, Nikus K, et al. 2021. GRINL1A complex transcription unit containing GCOM1, MYZAP, and POLR2M genes associates with fully penetrant recessive dilated cardiomyopathy. Front Genet 12: 2358. 10.3389/fgene.2021.786705 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases